GMOD
Overview
… formerly titled “GMOD for the Biologist”.
This page provides an overview of the GMOD project. It does not assume any particular background in computing.
Contents
- 1 Introduction
- 2 What is a GMOD?
- 3 Technologies
- 4 The Components of GMOD
- 5 Case Studies
- 6 Computing
Introduction
With the amount of technical documentation available for GMOD, the casual observer would be forgiven if they concluded that GMOD was a project about software. But it’s not: GMOD has been created for biologists and in the real world, it is used by biologists. However, the creators of GMOD are mostly not practicing biologists and the look and the feel of most GMOD documentation reflects this. What we will attempt to do is discuss GMOD from the researchers’ perspective. This does not simply mean describe what the software does. If you look, for example, at a typical GBrowse page (e.g. GBrowse view of human chromosome 7), you’ll understand immediately what GBrowse is built to do, and a few more minutes of clicking and scrolling will reveal all sorts of useful ways to display and query the data. A modern biologist knows a great deal about bioinformatics functionality already. This introduction is more concerned with answering practical questions, such as given the data I have, what database should I use?”, “do I even need a database?, or how hard is this going to be?.
In our experience we find that most biologists want to focus on the science. They may have little knowledge of programming languages or databases, and only passing interest in the IT minutiae. They have deep knowledge of their own data and know how such data can be viewed and analyzed. These biologists want to know how to efficiently create a useful set of tools for their data, and to be assured that their platform and tools can be easily maintained in an environment where resources may be limited. We will attempt to address these sorts of questions.
By the way, the word we used here refers to the GMOD Help Desk. The Help Desk is a good resource for biologists who want to learn more about GMOD. Feel free to email us at help@gmod.org.
What is a GMOD?
GMOD is a collection of interconnected applications and databases that biologists use as repositories and as tools. That connectivity is really the key here. Bioinformatics applications and databases are produced at a steady rate and this output is described each month in a number of different journals. There’s no lack of tools, but many of these tools will be little used as prospective users may not have the resources or expertise required to install the tool and hook it up to their data. What is generally lacking is a concerted effort to produce tools and databases that will work together; GMOD fills this void by providing the means to store data and a comprehensive set of tools for manipulating that data.
GMOD also describes a community. Many of the GMOD software components are mature software with many human-years of software development behind them. The design, development, and testing has been driven by a diverse group of software developers, scientists, and laboratories that use or improve these software components every day. The demand for software like this has been strong since genome sequence started to appear and many of the first genome databases used GMOD components. GMOD database and software components have developed and expanded with the massive growth and development of genome projects and the changing needs of users.
GMOD is also that specific thing that is installed on your computer. It may be the private viewer to your latest data that a student set up over the weekend. It may a terabyte-size database and suite of web applications developed over many years at a central laboratory. It may a database of experimental data accessible by script, or it may the annotation tool that you use to describe your favorite genome.
GMOD does not claim to cover every potential data storage, analysis or manipulation request that a biologist may have, but GMOD is a project directed by its user base: the biologists lead and the software developers follow, not the other way around. If you find your predicament is not addressed, or is only partially addressed, by what’s available in GMOD, your first step should be to contact the GMOD Help Desk or one of the main mailing lists like gmod-announce to make sure that your understanding of the available GMOD resources is correct. When you’re in touch with some knowledgable person, try to get a sense of what the solution might be, or its degree of difficulty. It may be that your solution may entail something simple, or it may be that a project may have to be created, complete with partnerships and grants. GMOD participants are always interested in seeing GMOD take off in new directions.
Is It Just for Model Organisms?
GMOD stands for Generic Model Organism Database; it was named back in the days when there were a handful of model organisms and it appeared that obtaining the genomic sequence of an organism was a prohibitively expensive proposition, taking months or years to accomplish. With the ease and speed at which genomes can now be sequenced, few scientists would consider their organism a ‘model’ in this early sense of the word, so we suggest users think of the M as standing for Myriad or My.
Any organism with any kind of sequence associated with it is a good candidate as a subject for a GMOD database. There are GMOD databases with just protein sequence in them like the S. cerevisiae Proteome Browser. There are GMOD databases with EST sequence only, such as the Cattle EST Gene Family Database. There are GMOD databases that are concerned primarily with gene expression, such as the Emiliania huxleyi Serial Analysis of Gene Expression database. We even find GMOD databases dedicated to collections of RNA sequence like the Leishmania tarentolae RNA Editing database. We have also heard of GMOD databases for oligonucleotides and plasmids. See GMOD Users for a list of other examples. The list of GMOD databases demonstrates that GMOD is widely used, with many organisms represented, and that these databases can hold sequences of any kind.
Technologies
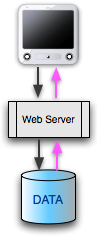 |
Most GMOD installations have a general architecture in common. There
is a data source: the database. This does not have to be a relational
database; it could be a file or a set of files with or without some kind
of index. There's a lot of flexibility at the data level. Choosing this
database and loading it will be tasks you'll give a lot of thought to.
|
The Components of GMOD
GMOD is made up databases, applications, and adaptor software that connects these components together. Some of the most popular packages are discussed below.
What is GBrowse?
GBrowse is short for Genome Browser, or Generic Genome Browser. GBrowse is probably GMOD’s most popular component and almost all of the databases listed in GMOD Users use GBrowse. It is fairly easy to install, requiring only basic command-line familiarity. GBrowse is popular is because it is a supremely capable browser. The picture below is a partial screenshot of a GBrowse page taken from the Human chromosome 7 database at TCAG. A bit of jargon: the rows, each depicting one sort of data, are called tracks and tracks are populated by one or more images called glyphs, plus text.
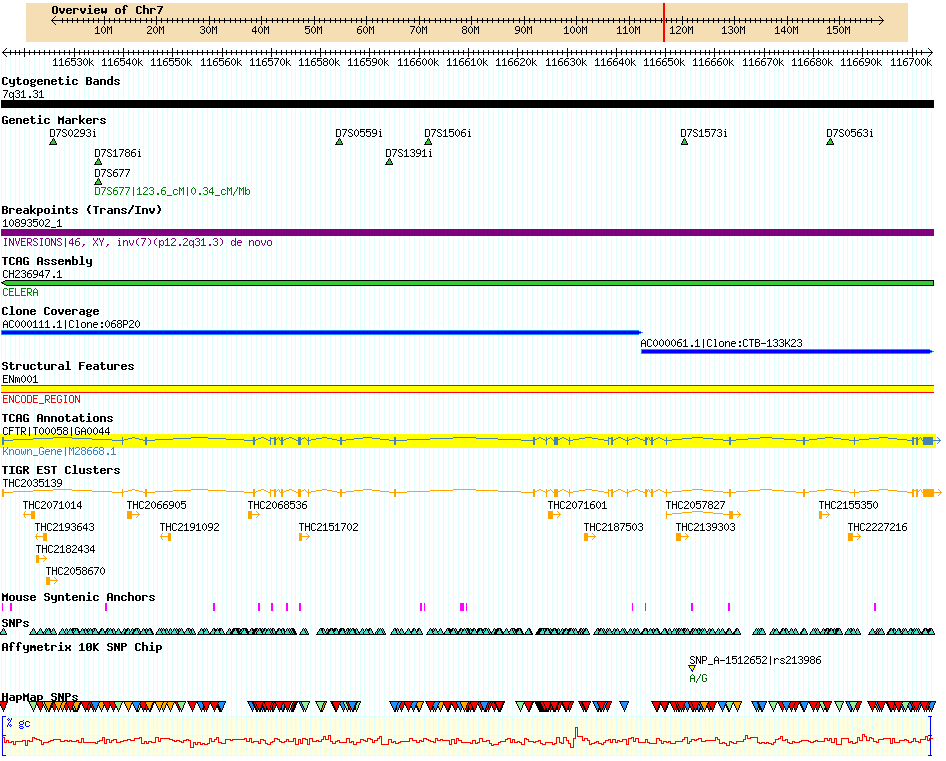
GBrowse comes with a large library of glyphs, including pie charts, dot plots, histograms, and X-Y plots suitable for quantitative data, as well as the expected array of glyphs that describe sequences and sequence annotation. It is also highly configurable, meaning you can do quite a bit of customization of the glyphs, you can link glyphs to URLs of your choice, you can internationalize the application to display different languages, you can connect and retrieve data from any database, and more. This sort of work generally requires either modifying GBrowse’s configuration files or adding your own code. GBrowse is written in Perl; as GBrowse is designed to be customized, extending its functionality with your own code should not require expert coding skill.
JBrowse
JBrowse is a genome browser with a fully dynamic HTML5 user interface, being developed as the successor to GBrowse. It is very fast and scales well to large datasets. JBrowse is javascript-based and does almost all of its work directly in the user’s web browser, with minimal requirements for the server. JBrowse’s features include:
- Fast, smooth scrolling and zooming. Explore your genome with unparalleled speed.
- Scales easily to multi-gigabase genomes and deep-coverage sequencing.
- Supports GFF3, BED, FASTA, Wiggle, BigWig, BAM, VCF (with tabix), REST, and more. BAM, BigWig, and VCF data are displayed directly from the compressed binary file with no conversion needed.
- Very light server resource requirements. JBrowse has no back-end server code, just tools for formatting data files to be read directly over HTTP. Serve huge datasets from a single low-cost cloud instance.
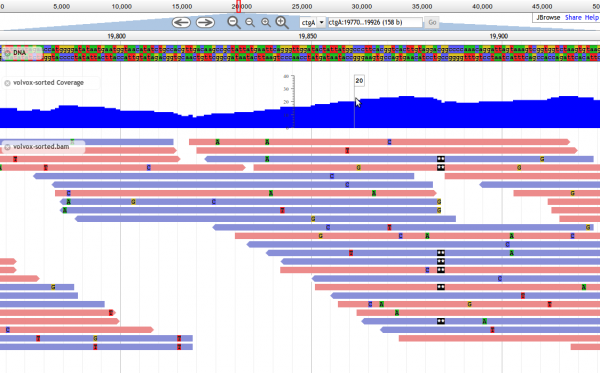
Screenshot of JBrowse in action
Relational Databases
For those unfamiliar with databases, the brief guide to databases provides a gentle introduction.
Relational databases are today’s tool of choice when faced with the problem of storing complex or multifaceted data, assuming that the data is, or can be, broken down into ever smaller bits of data. All atomized data will end up in one field, analogous to the way that data can be organized as columns in a spreadsheet. Fields describing aspects of a “thing” (an entity) are organized together into tables. For example, a relational database may have a table called gene with gene.name and gene.geneid fields and a protein table with protein.name, protein.proteinid and protein.sequence fields.
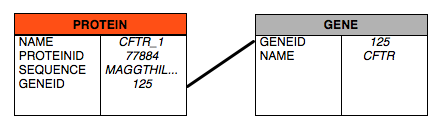
The picture above shows these two tables, and explains the term relational. The relation between the tables is the shared geneid - we add the geneid field to the protein table to indicate that the CFTR_1 protein record relates back to a specific gene in the gene table. This geneid field in protein, which originates in gene and whose values are stored in gene, is an example of a foreign key, a field from a table that is shared by one or more other tables.
For a given collection of data, genomic sequence and annotation for example, there will be more than one way to represent the data relationally. A given relational design, essentially tables and fields, is called a schema (think of the schema as a blueprint, empty, and the schema populated with data as the database). Both Chado and BioSQL can store genomic data for example, but they do it differently. The details of schema design are not relevant here but one can say that the designer may think about some of these general concerns:
- The degree of data abstraction, which is related to a database concept called normalization and to the flexibility of the schema
- The legibility of the schema, which has to do with the ease of using it
- The breadth of the schema, in terms of the data types it could contain
From the scientific perspective one can ask related questions:
- How flexible is a given schema?
- Can it handle my data now and in the future?
- Will using a given schema be easier or harder to use than some other schema?
This last question relates mostly to the degree of abstraction of the schema, not to the actual programming languages used.
All of today’s relational databases are created and loaded and queried using one language, SQL; programmers use their chosen language (Perl, Java, Python, etc.) to execute SQL queries and process the results.
See also:
Chado and BioSQL
So when you choose to use a relational schema it will all really come down to you and your data, not technical details. Chado is one of the relational databases that are used in GMOD, the other being BioSQL.
The differences are clear. BioSQL is quite focussed and is concerned with:
- Sequence
- Sequence annotation
- Phylogeny
- Publications
It is also a thoroughly modern schema in that it uses OBO-style ontologies, such as the Gene Ontology (GO). This is a requirement, given the ubiquity of ontologies and their ability to describe and organize data.
Chado’s focus is broader. Its tables are broken down into groups called modules; the modules are the following:
- Audit - for database audit trails
- Companalysis - for data from computational analysis
- Contact - for people, groups, and organizations
- Controlled Vocabulary (cv) - for controlled vocabularies and ontologies
- Expression - for summaries of RNA and protein expresssion
- General - for identifiers
- Genetic - for genetic data and genotypes
- Library - for descriptions of molecular libraries
- Mage - for microarray data
- Map - for maps without sequence
- Natural Diversity (ND) - for multiple experiments, such as phenotyping and genotyping
- Organism - for taxonomic data
- Phenotype - for phenotypic data
- Phylogeny - for organisms and phylogenetic trees
- Publication (pub) - for publications and references
- Sequence - for sequences and sequence features
- Stock - for specimens and biological collections
- WWW -
It is also possible to add modules to Chado. For instance, in early 2007 a module called mage was added to addresses microarray data. Other possibilities that are being discussed are modules for ecological data and additional work for phenotypic data, extending the existing phenotype module. The real point is that Chado has been designed to allow extensibility, and one can either formally propose that Chado acquire some new functionality as a module or you can add tables to Chado in the privacy of your own server.
Chado is also ontology-aware. One could state this even more forcefully: Chado depends on ontologies. For example, in Chado’s Sequence module it is expected that all stored sequences are identified by one or more terms from the Sequence Ontology. A quick scan of the tables in Chado, more than 100, shows that about half of the tables contain the field, foreign key, cvterm, referring to an ontology term. The ontology used as source for a term could be one of many but people in the field tend to rely on OBO ontologies. So the ontology could be a common and general one like GO, the Gene Ontology, or something highly specific to a group of organisms like the Drosophila Anatomy ontology or the Mammalian Phenotype ontology. In conjunction with Chado, these ontologies give you a database that is extremely flexible, and as your ontologies expand, so does the expressive capability of the system.
There is a cost to this flexibility and breadth: Chado is complex and it is unlikely that someone unfamiliar with Chado would be able to install it and then immediately set about loading it with biological data of different sorts. Fortunately there are mailing lists you can contact, as well as the GMOD Help Desk, and number of pages on this Wiki discussing Chado (see Getting Started with Chado and the Chado Manual).
GFF Databases
In addition to relational database schemas like Chado and BioSQL you will also encounter GFF databases. GFF is a compact format for describing sequence and sequence annotations. GMOD installations like the Human Chromosome 1 database described above are concerned solely with sequence and annotation and the entire contents of such a database can be represented as GFF. For small installations the entire database can be just a set of GFF text files (in fact, you can install GBrowse on your personal computer and then browse Saccharomyces and Volvox genomic sequence, reading directly from GFF files installed along with GBrowse - try it!). But when the amount of GFF gets too large to be read into memory all at once you have to store the GFF in some form that’s indexed for fast retrieval. The solution is to load the GFF into MySQL or some other sort of database management system, this assures good performance even if you have very large amounts of data in GFF format. This is accomplished by using the Bio::DB::GFF or Bio::DB::SeqFeature GBrowse Adaptors.
What are WebApollo and Apollo?
Unlike JBrowse and GBrowse, which only function as a sequence browser, WebApollo and its standalone predecessor Apollo, are for both viewing and manually annotating genomes. WebApollo is a plugin for JBrowse that allows multiple users to annotate genomes concurrently. Changes made by others are automatically and immediately updated in the user’s browser window, ensuring that there is no duplication of effort, and allowing several users to annotate parts of the same sequence at the same time. A full history of all edits is kept, and the changes made in editing sessions can be approved or rejected by an administrator before being saved. WebApollo shares JBrowse’s fast, flexible browsing interface, and users require only a web browser to use it.
Apollo is a standalone Java application for manual sequence annotation, and is the predecessor of WebApollo. Apollo can read and write to Chado databases, but lacks the instant updates that WebApollo features. We recommend using WebApollo as it is under active development and has a more full feature set than Apollo.
What are MAKER and DIYA?
GBrowse and Apollo both deal with genome annotations, but where do these annotations come from? Frequently they come from a genome annotation pipeline, a software package or series of software packages that take an assembly (and other things) as input and produces an annotated genome, often with gene models, ESTs, proteins, and almost anything else that can be tied back to a genomic sequence.
MAKER is a genome annotation pipeline that produces annotated eukaryotic genomes, and DIYA is a genome annotation pipeline for prokaryotic genomes (and both do more than that too). They both produce gene models in GFF, a file format that can be directly loaded into GBrowse, Apollo, and Chado.
What is Pathway Tools?
Pathway Tools is a software system for creating organism-specific databases. It contains extensive functionality that spans from genomes to pathways including a genome browser, metabolic pathway predictor and viewer, and regulatory network viewer, as well as a large number of interactive annotation tools.
What is CMap?
CMap is a popular comparative map viewer. It was initially created for use at Gramene but was redesigned to be used for any organism or set of organisms. It can display genetic maps or physical maps and draw the relations between the two. It will also show synteny. It is written in Perl and requires an underlying RDBMS such as MySQL. If you need to display maps or syntenic relationships, you may need more than GBrowse.
And SynView? or Sybil? or GBrowse_Syn?
Yes, there are other comparative genomics viewers. The alternatives to CMap are GBrowse_syn, Sybil, and SynView. Sybil stores its data in Chado and accommodates a variety of different analyses; go to the Sybil Web site if you want to learn more. GBrowse_syn and SynView build upon GBrowse, and they can be considered a bit simpler than Sybil and CMap. More information is available on their websites to help you determine which is most suitable for you.
What is Tripal?
Tripal is a web frontend for a Chado database that provides both an attractive, slick website for accessing and disseminating Chado data, and an interface for local users to upload and edit data in the database. Tripal is based on the popular content management system Drupal, and creates a customisable website from the Chado database. Tripal includes a number of analysis modules that allow the incorporation of external data (for example, Gene Ontology annotations), and tools such as GBrowse, Galaxy, and CMap can be integrated into the site. Tripal is very customisable and, as it is based on Drupal, extra website content can easily be added using the standard Drupal functionality. For groups looking to avoid the substantial investment of time and effort involved in creating a website for data display and dissemination, Tripal offers a simple, stylish solution.
What is Modware?
Modware is a middleware package used in GMOD, written in Perl. Middleware is software that mediates the exchange of information between applications, e.g. between web pages and databases; please see the GMOD Middleware page for technical details. GMOD developers have evaluated a number of Perl middleware packages and decided that Modware is best suited to GMOD Perl development. Like Bioperl, Modware may be a package that you may need to install but won’t need to understand in any detail.
What is BioPerl?
BioPerl is a popular bioinformatics toolkit written in Perl. The reason we mention it here is because many of the GMOD Components use parts of it. You will not have to learn BioPerl in order to use GMOD but you may have to install it.
BioPerl offers some attractive ways to store genomic data without requiring a relational database. We discussed Chado and BioSQL above; these two relational schemas require the prior installation of some free, open source RDBMS like MySQL or PostgreSQL. Installing these pieces, schema plus RDBMS, is not necessarily difficult, but if you only have sequence and sequence annotation, you can set up a sequence or genome browser using BioPerl, GBrowse, and an Apache web server. You can use either the Bio::DB::GFF module from BioPerl or the Bio::DB::SeqFeature module. See A Simple Sequence Browser below.
And What Else is in GMOD?
A number of other software packages, listed below, classified by general function. One might be tempted to think of this as a shopping list, choosing one of each. But it may also be useful to think of what is absolutely essential first and consider these other components as add-ons. Some of these components are only loosely coupled to some of the more core components described above; an application might use its own methods to store data and not use Chado. Or, a component may be written in Java and not Perl, so it would not be able to communicate with a Perl application. For something to be considered a GMOD component it does not, at this time, have to connect to some other component.
Community Annotation Wiki TableEdit
|
Gene Expression Visualization
|
Molecular Pathway Visualization
Bio%253A%253AChado%253A%253ASchema
|
Case Studies
What we are attempting to do here is anticipate some of the basic requirements of the scientist. The classic situation is that he or she has data of some type, or of many different types, and needs to set up both a data repository and a viewer on this data. We are assuming that the scientist is not a programmer or an IT expert but that he or she is willing to learn the necessary skills or has a student available to do the required work.
A Simple Sequence Browser
- The data: sequence (genomic DNA or ESTs or proteins or cDNAs or some combination of these or …)
- The goal: create a browser to query and view sequence and sequence annotations
- The core software: GBrowse, Apache Web server, and Bioperl
- The hardware: a server running Unix (Linux or Mac) or Windows
- Figure out what the annotations should be (i.e. gene coordinates, motif matches, oligonucleotide matches, etc. You can try using annotation pipelines like MAKER to automatically build these.)
- Install core software
- Create or gather the annotations (BLAST results or HMMER results or GenBank files or …)
- Transform all the annotations into a format suitable for loading (GFF format)
- Load GFF into the GFF database
- Configure GBrowse
Possible challenge: Step 4, converting all the annotations to GFF (scripts may available to perform all the conversions, or you may have to write some of the conversion code yourselves)
- Skills needed: basic command-line competence, perhaps basic Perl competence if you have to write any custom conversion code
- Resources available: documentation at GMOD.org, the GMOD Help Desk, the GMOD Mailing Lists
Recommendation
Highly recommended. Setting this up will give you a good sense of how the software pieces interoperate. Not only that, but GBrowse is fun and it comes with sample databases so once it’s installed you have actual genome sequence to play with. You can even get GBrowse running nicely on a laptop.
A Simple Sequence Browser plus a Sequence Annotator
- The data: sequence (genomic DNA or ESTs or cDNAs or some combination of these or …)
- The goal: create a browser to query and view sequence and sequence annotations along with an editor to manually annotate the sequences
- The core software: GBrowse, Apollo, Chado (plus relational database), Apache Web server, and BioPerl
- The hardware: a server running Unix (Linux or Mac) or Windows
- Figure out what the annotations should be (gene coordinates or motif matches or oligonucleotide matches or hand-made annotations or some combination of these or …)
- Install core software
- Create or gather the annotations (BLAST results or HMMER results or GenBank files or …)
- Transform all the annotations into a format suitable for loading (GFF format)
- Load GFF into the Chado database
- Install and configure Gbrowse
- Install and configure Apollo
A challenge: Step 2, installing core software (with more components you have a more complex system and more potential pitfalls, and Chado and its relational database is a fairly detailed install)
Possible challenge: Step 4, converting all the annotations to GFF (scripts may available to perform all the conversions, or you may have to write some of the conversion code yourselves)
Skills needed: basic command-line competence, perhaps basic Perl competence if you have to write any custom conversion code. Some understanding of relational databases for the Chado installation. Basic Java competence for the Apollo installation.
Resources available: documentation at www.gmod.org, the GMOD Help Desk, the GMOD mailing lists
Recommendation
If you’re a GMOD novice, install GBrowse by itself first (A Simple Sequence Browser), then consider this system.
A Browser for a Stock Collection
- The data: the stock collection data in some structured form (Excel or Word or …)
- The goal: create a browser to query and view your laboratory’s stock collection
- The core software: Chado (and its relational database), Apache Web server, and Turnkey
- The hardware: a server running the Unix (Linux or Mac) or Windows operating system.
- Install core software
- Load stock collection data into the Chado database
- Create a Tripal-based Web site
Challenges:
- Possible challenge: Step 1, installing core software (Chado and its relational database is a fairly detailed install)
- A challenge: Step 2, loading the stock collection data into Chado (scripts will not be available to perform this loading, you will have to create the code yourselves)
- Possible challenge: Step 2, loading the data. The Chado schema may not be properly configured for your data and may need to be modified.
- Possible challenge: Step 3, running Turnkey to automatically create your browser. Turnkey is a new tool. It has been used successfully in testing and at ParameciumDB but not all possibilities have been tested.
Skills needed: General IT expertise (Turnkey automates the creation of Web sites but it is an expert’s tool) Basic programming competence to write the custom conversion code.
Resources available: documentation at www.gmod.org, the GMOD Help Desk, the GMOD Mailing Lists.
Recommendation
Consider whether you want to explore uncharted territory or not. Could be fairly straightforward for the expert, or could be challenging.
A Browser for Microarray Data
- The data: microarray data in Affymetrix format
- The goal: create a browser to query and view your laboratory’s microarray
- The core software: Chado, Apache Web server, and …
- The hardware: a server running Unix (Linux or Mac) or Windows
Challenge: Chado can hold the microarray data using its Mage module and applications exist to view raw microarray data (e.g. Caryoscope, GeneXplorer) but these applications don’t connect to Chado.
Resources available: documentation at www.gmod.org, the GMOD Help Desk, the GMOD mailing lists
Recommendation
Either wait for the connectors to be built to some application or form a partnership with GMOD scientists and developers to see that the connectors are built.
A Browser for Map Data
- The data: map data (genetic map data or physical map data or visual map data or some combination of these)
- The goal: create a browser to query and view your maps, within a species or across species
- The core software: GBrowse, Apache Web server, and CMap or SynView or Sybil.
- The hardware: a server running Unix (Linux or Mac) or Windows
- Choose the right map software, based on your map data and resources.
- Install core software.
- Load map data.
Possible challenge: Step 2, the installation. This may tricky if you choose one of the more fully featured packages (CMap or Sybil).
Possible challenge: Step 3, the loading. It is likely that some custom coding would be required since map data comes in all sorts of different forms.
Skills needed: Basic command-line competence. Some understanding of relational databases for CMap or Sybil. Basic programming competence to write the custom loading code.
Resources available: documentation at www.gmod.org, the GMOD Help Desk, the GMOD mailing lists
Recommendation
Choose one. GMOD offers good choices here, it comes down to your data and your resources. SynView is the easiest, and it comes with GBrowse.
Computing
Personnel, Hardware and Operating System
Computing Requirements discusses the personnel, hardware, and operating system requirements and choices for implementing GMOD components.
Software
GMOD software relies on other software to function. This section lists some other key open source packages that you may need.
Databases
The Relational Databases section above introduced many relational database concepts. Databases and GMOD discuses database management system choices in GMOD. It also introduces some additional terminology.
Programming Languages
Two programming languages are popular in GMOD: Perl and Java. For most tasks you won’t need to do any programming in either language. You will just need to know how to install these languages and how to install programs written in these languages. See Computing Requirements for more.
Perl
The programming language most used in the bioinformatics realm. Also the language most used by GMOD developers. It is well-suited to text and data processing and is also characterized by an extensive open source library, so it’s highly functional. Many of GMOD components use BioPerl, a bioinformatics toolkit written in Perl.
Some pieces of GMOD, like GBrowse, can be extended or customized using Perl but beginners’ skills in Perl would be sufficient for this work. Just installing and using GBrowse in a conventional way does not require knowledge of Perl or BioPerl.
Java
Java is arguably the world’s most popular programming language but it is not as popular for command-line work on Unix as Perl. It’s encountered in GMOD primarily as a language to construct user interfaces (e.g. Apollo).
Apache, the Web Server
If you want to set up an application that displays web pages, you will need a web server on your computer. If you don’t already have one installed, you will want to use the Apache Web server (also known as the Apache HTTP Server), which is free, fast, secure, and reasonably simple to install on Unix or Windows.
Glossary
The GMOD Glossary explains many terms related to GMOD, bioinformatics, and the computing technologies used in GMOD.
Licenses
Most GMOD Components have no restrictions on using them. Those few components that do impose restrictions will clearly state that they have restricted licenses.