GMOD
WebApollo Tutorial 2012
This WebApollo tutorial was presented by Ed Lee at the 2012 GMOD Summer School.
To follow along with the tutorial, you will need to use AMI ID: ami-a1de69c8, name: GMOD 2012 start day 3, available in the US East (N. Virginia) region. See the GMOD Cloud Tutorial for information on how to get this AMI.
Please note that WebApollo was not officially released when this session was taught, and this tutorial was run using a beta version of the tool.
Contents
- 1 Introduction
- 2 Using WebApollo
- 3 Installing WebApollo
- 4 Installation
- 5 Deploy servlet
- 6 Configuration
- 7 Data generation
Introduction
Using WebApollo
WebApollo is a web-based application, so the only requirement to use it is a web browser. It has been tested with Chrome, Firefox, and Safari. It currently does not support Internet Explorer.
A WebApollo demo with the Pythium data has been set up on the virtual machine. We’ll use the demo to view WebApollo’s functionality.
Important: before you load the demo, you’ll need to do a few things:
$ cd /usr/local/tomcat/tomcat7/webapps/WebApolloDemo/jbrowse
$ sudo wget http://icebox.lbl.gov/webapollo/data/genome.css
$ sudo mv genome.css.1 genome.css
Next we need to modify data/bam_trackList.json. You’ll want to modify
the data_url and index_url elements and add a sourceUrl element.
This is how it currently looks:
"data_url" : "http://ec2-##-##-##-##.compute-1.amazonaws.com:8080/WebApolloDemo/jbrowse/data/bam/scf1117875582023.bam",
"index_url" : "http://ec2-##-##-##-##.compute-1.amazonaws.com:8080/WebApolloDemo/jbrowse/data/bam/scf1117875582023.bam.bai",
We want to change it to:
"data_url" : "bam/scf1117875582023.bam",
"index_url" : "bam/scf1117875582023.bam.bai",
"sourceUrl" : "data/",
Point your browser to
http://ec2-##-##-##-##.compute-1.amazonaws.com:8080/WebApolloDemo.
(e.g.,
http://ec2-184-73-92-243.compute-1.amazonaws.com:8080/WebApolloDemo).
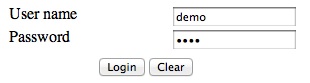
The user name and password are both demo.
![]()
Click on the Edit annotations button.
![]()
We only have one contig to work with. Click on the Edit button.
- Annotation track
- Add evidence tracks (maker, snap_masked, blastn, blastx, est2genome, protein2genome, BAM alignments)
- Moving around the contig
- Go to region
scf1117875582023:629476..633770 - Selection
- Edge matching
- Create annotation
- Drag-n-drop
- Delete exon (notice the change in CDS)
- Add exon
- Non-canonical splice sites (GT / AG)
- Zoom to base
- DNA track
- Highlighting
- 6-frame translation
- DNA sequence insertion, deletion, substitution
- Zoom back out
- Change exon boundaries
- Merge (exons, transcripts)
- Split (exons, transcripts)
- Make intron
- Duplicate
- Set translation start
- Realtime client updating
- Undo / Redo
- Comments
- DBXrefs
- Get sequence
- Sequence searching
Installing WebApollo
Server operating system
Any Unix like system (e.g., Unix, Linux, Mac OS X)
Prerequisites
- Servlet container (must support servlet spec 3.0+) [officially supported: Tomcat 7]
- Relational Database Management System [officially supported: PostgreSQL]
- User database helper scripts
- Perl modules
- DBI
- DBD module for your specific RDBMS [officially supported: DBD::Pg for PostgresSQL]
- Perl modules
- Data generation pipeline (see JBrowse
prerequisites for more information
on its prerequisites)
- Perl modules
- BioPerl 1.6
- JSON
- JSON::XS (optional, for speed)
- PerlIO::gzip
- Heap::Simple
- Heap::Simple::XS
- Devel::Size
- Bio::GFF3::LowLevel::Parser
- System packages
- libpng12-0
- libpng12-dev
- Perl modules
- Sequence search (optional)
- Blat (along with a configured search database)
Note: All prerequisites have already been installed on the course’s machine
Installation
The installation steps will be done in the command line. SSH into your AWS machine.
Uncompress the WebApollo.tgz tarball.
$ cd
$ tar -xvzf WebApollo.tgz
User database
First we’ll need to create a database that will contain user permission
information. WebApollo uses this database to determine who can access
and edit annotations for a given sequence. We’ll create the user
web_apollo_users_admin to be the owner of the database with the
password web_apollo_users_admin (enter it when prompted). The user
will not be a superuser nor will it be able to create new roles. But it
will be able to create databases.
$ createuser -P web_apollo_users_admin
Enter password for new role:
Enter it again:
Shall the new role be a superuser? (y/n) n
Shall the new role be allowed to create databases? (y/n) y
Shall the new role be allowed to create more new roles? (y/n) n
Let’s now create the database. We can call it whatever we want, so let’s
call it web_apollo_users.
$ createdb -U web_apollo_users_admin web_apollo_users
Now that the database is created, we need to load the schema to it.
$ cd ~/WebApollo/tools/user
$ psql -U web_apollo_users_admin web_apollo_users < user_database_postgresql.sql
Now the user database has been setup.
Let’s populate the database.
First we’ll create an user with access to WebApollo. We’ll use the
add_user.pl script in ~/WebApollo/tools/user. Let’s create an user
named web_apollo_admin with the password web_apollo_admin.
$ ./add_user.pl -D web_apollo_users -U web_apollo_users_admin -P web_apollo_users_admin \
-u web_apollo_admin -p web_apollo_admin
Next we’ll add the genomic sequence ids for our organism in the
database. We’ll use the add_tracks.pl script in the same directory. We
need to generate a file of genomic sequence ids for the script. For
convenience, there’s a script called extract_seqids_from_fasta.pl in
the same directory which will go through a FASTA file and extract all
the ids from the deflines. We’ll use the data from
MAKER. Let’s first create the list of genomic
sequence ids. We’ll store it in ~/scratch/seqids.txt. We’ll want to
add the prefix “Annotations-” to each identifier.
$ mkdir ~/scratch
$ ./extract_seqids_from_fasta.pl -p Annotations- -i ~/maker_output/scf1117875582023.fa \
-o ~/scratch/seqids.txt
Now we’ll add those ids to the user database.
$ ./add_tracks.pl -D web_apollo_users -U web_apollo_users_admin -P web_apollo_users_admin \
-t ~/scratch/seqids.txt
Now that we have an user created and the genomic sequence ids loaded,
we’ll need to give the user permissions to access the sequence. We’ll
have the user have read, write, and user manager permissions. We’ll use
the set_track_permissions.pl script in the same directory. We’ll need
to provide the script a list of genomic sequence ids, like in the
previous step.
$ ./set_track_permissions.pl -D web_apollo_users -U web_apollo_users_admin \
-P web_apollo_users_admin -u web_apollo_admin -t ~/scratch/seqids.txt -r -w -m
We’re all done setting up the user database.
Note that we’re only using a subset of the options for all the scripts mentioned above. You can get more detailed information on any given script (and other available options) using the “-h” or “–help” flag when running the script.
Deploy servlet
We need to deploy the WAR file in the war directory from the unpacked
tarball. We need to go into the webapps directory in our Tomcat
installation. Tomcat’s installed in /usr/local/tomcat/tomcat7/.
$ cd /usr/local/tomcat/tomcat7/webapps
Next we need to create the directory that will contain the application.
Note that you’ll need to use sudo for this, since only root has access
to the directory.
$ sudo mkdir WebApollo
Now we’ll go into the newly created directory and unpack the WAR file into it.
$ cd WebApollo
$ sudo jar -xvf ~/WebApollo/war/WebApollo.war
That’s it! We’re done installing WebApollo. Now we need to move on to configuring the application.
Configuration
Most configuration files will reside in
/usr/local/tomcat/tomcat7/webapps/WebApollo/config. We’ll need to
configure a number of things before we can get WebApollo up and running.
Main configuration
The main configuration is stored in
/usr/local/tomcat/tomcat7/webapps/WebApollo/config/config.xml. Let’s
take a look at the file.
<?xml version="1.0" encoding="UTF-8"?>
<server_configuration>
<!-- mapping configuration for GBOL data structures -->
<gbol_mapping>/config/mapping.xml</gbol_mapping>
<!-- directory where JE database will be created -->
<datastore_directory>ENTER_DATASTORE_DIRECTORY_HERE</datastore_directory>
<!-- minimum size for introns created -->
<default_minimum_intron_size>1</default_minimum_intron_size>
<!-- size of history for each feature - setting to 0 means unlimited history -->
<history_size>0</history_size>
<!-- overlapping strategy for adding transcripts to genes -->
<overlapper_class>org.bbop.apollo.web.overlap.OrfOverlapper</overlapper_class>
<!-- class for comparing track names (used for sorting in lists) -->
<track_name_comparator_class>org.bbop.apollo.web.track.DefaultTrackNameComparator</track_name_comparator_class>
<!-- user authentication/permission configuration -->
<user>
<!-- database configuration -->
<database>
<!-- driver for user database -->
<driver>org.postgresql.Driver</driver>
<!-- JDBC URL for user database -->
<url>ENTER_USER_DATABASE_JDBC_URL</url>
<!-- username for user database -->
<username>ENTER_USER_DATABASE_USERNAME</username>
<!-- password for user database -->
<password>ENTER_USER_DATABASE_PASSWORD</password>
</database>
<!-- class for generating user authentication page (login page) -->
<authentication_class>org.bbop.apollo.web.user.localdb.LocalDbUserAuthentication</authentication_class>
</user>
<tracks>
<!-- path to JBrowse refSeqs.json file -->
<refseqs>ENTER_PATH_TO_REFSEQS_JSON_FILE</refseqs>
<!-- annotation track name the current convention is to append
the genomic region id to the the name of the annotation track
e.g., if the annotation track is called "Annotations" and the
genomic region is chr2L, the track name will be
"Annotations-chr2L".-->
<annotation_track_name>Annotations</annotation_track_name>
<!-- organism being annotated -->
<organism>ENTER_ORGANISM</organism>
<!-- CV term for the genomic sequences - should be in the form
of "CV:TERM". This applies to all sequences -->
<sequence_type>ENTER_CVTERM_FOR_SEQUENCE</sequence_type>
</tracks>
<!-- path to file containing canned comments XML -->
<canned_comments>/config/canned_comments.xml</canned_comments>
<!-- tool to be used for sequence searching. This is optional.
If this is not setup, WebApollo will not have sequence search support -->
<sequence_search_tool>
<!-- class for handling search -->
<class>org.bbop.apollo.tools.seq.search.blat.BlatCommandLine</class>
<!-- configuration for search tool -->
<config>/config/blat_config.xml</config>
</sequence_search_tool>
</server_configuration>
Let’s look through each element in more detail with values filled in.
<!-- mapping configuration for GBOL data structures -->
<gbol_mapping>/config/mapping.xml</gbol_mapping>
File that contains type mappings used by the underlying data model. It’s best not to change the default option.
<!-- directory where JE database will be created -->
<datastore_directory>/data/dataHome/web_apollo/annotations</datastore_directory>
Directory where user generated annotations will be stored. The data is
stored using Berkeley DB. We’ll use
/data/dataHome/web_apollo/annotations.
<!-- minimum size for introns created -->
<default_minimum_intron_size>1</default_minimum_intron_size>
Minimum length of intron to be created when using the “Make intron” operation. The operation will try to make the shortest intron that’s at least as long as this parameter. So if you set it to a value of “40”, then all calculated introns will be at least of length 40.
<!-- size of history for each feature - setting to 0 means unlimited history -->
<history_size>0</history_size>
The size of your history stack, meaning how many “Undo/Redo” steps you can do. The larger the number, the larger the storage space needed. Setting it to “0” makes it to that there’s no limit.
<!-- overlapping strategy for adding transcripts to genes -->
<overlapper_class>org.bbop.apollo.web.overlap.OrfOverlapper</overlapper_class>
Defines the strategy to be used for deciding whether overlapping
transcripts should be considered splice variants to the same gene. This
points to a Java class implementing the
org.bbop.apollo.overlap.Overlapper interface. This allows you to
create your own custom overlapping strategy should the need arise.
Currently available options are:
- org.bbop.apollo.web.overlap.NoOverlapper
- No transcripts should be considered splice variants, regardless of overlap.
- org.bbop.apollo.web.overlap.SimpleOverlapper
- Any overlapping of transcripts will cause them to be part of the same gene
- org.bbop.apollo.web.overlap.OrfOverlapper
- Only transcripts that overlap within the coding region and within frame are considered part of the same gene
<!-- class for comparing track names (used for sorting in lists) -->
<track_name_comparator_class>org.bbop.apollo.web.track.DefaultTrackNameComparator</track_name_comparator_class>
Defines how to compare genomic sequence names for sorting purposes in
the genomic region selection list. Points to a class implementing the
org.bbop.apollo.web.track.TrackNameComparator interface. You can
implement your own class to allow whatever sorting you’d like for your
own organism. This doesn’t make much of a difference in our case since
we’re only dealing with one genomic region. The only available
implementation is:
- org.bbop.apollo.web.track.DefaultTrackNameComparator
- Sorts genomic sequence names lexicographically
Let’s take look at the “user” element, which handles configuration for user authentication and permission handling.
<!-- user authentication/permission configuration -->
<user>
<!-- database configuration -->
<database>
<!-- driver for user database -->
<driver>org.postgresql.Driver</driver>
<!-- JDBC URL for user database -->
<url>ENTER_USER_DATABASE_JDBC_URL</url>
<!-- username for user database -->
<username>ENTER_USER_DATABASE_USERNAME</username>
<!-- password for user database -->
<password>ENTER_USER_DATABASE_PASSWORD</password>
</database>
<!-- class for generating user authentication page (login page) -->
<authentication_class>org.bbop.apollo.web.user.localdb.LocalDbUserAuthentication</authentication_class>
</user>
Let’s first look at the database element that defines the database
that will handle user permissions (which we created previously).
<!-- driver for user database -->
<driver>org.postgresql.Driver</driver>
This should point the JDBC driver for communicating with the database. We’re using a PostgreSQL driver since that’s the database we’re using for user permission management.
<!-- JDBC URL for user database -->
<url>jdbc:postgresql://localhost/web_apollo_users</url>
JDBC URL to the user permission database. We’ll use
jdbc:postgresql://localhost/web_apollo_users since the database is
running in the same server as the annotation editing engine and we named
the database web_apollo_users.
<!-- username for user database -->
<username>web_apollo_users_admin</username>
User name that has read/write access to the user database. The user with
access to the user database has the user name web_apollo_users_admin.
<!-- password for user database -->
<password>web_apollo_users_admin</password>
Password to access user database. The user with access to the user database has the password </tt>web_apollo_users_admin</tt>.
Now let’s look at the other elements in the user element.
<!-- class for generating user authentication page (login page) -->
<authentication_class>org.bbop.apollo.web.user.localdb.LocalDbUserAuthentication</authentication_class>
Defines how user authentication is handled. This points to a class
implementing the org.bbop.apollo.web.user.UserAuthentication
interface. This allows you to implement any type of authentication you’d
like (e.g., LDAP). Currently available options are:
- org.bbop.apollo.web.user.localdb.LocalDbUserAuthentication
- Uses the user permission database to also store authentication information, meaning it stores user passwords in the database
- org.bbop.apollo.web.user.browserid.BrowserIdUserAuthentication
- Uses Mozilla’s BrowserID service for authentication. This has the benefits of offloading all authentication security to Mozilla and allows one account to have access to multiple resources (as long as they have BrowserID support). Being that the service is provided through Mozilla, it will require users to create a BrowserID account
Now let’s look at the configuration for accessing the annotation tracks for the genomic sequences.
<tracks>
<!-- path to JBrowse refSeqs.json file -->
<refseqs>ENTER_PATH_TO_REFSEQS_JSON_FILE</refseqs>
<!-- annotation track name the current convention is to append
the genomic region id to the the name of the annotation track
e.g., if the annotation track is called "Annotations" and the
genomic region is chr2L, the track name will be
"Annotations-chr2L".-->
<annotation_track_name>Annotations</annotation_track_name>
<!-- organism being annotated -->
<organism>ENTER_ORGANISM</organism>
<!-- CV term for the genomic sequences - should be in the form
of "CV:TERM". This applies to all sequences -->
<sequence_type>ENTER_CVTERM_FOR_SEQUENCE</sequence_type>
</tracks>
Let’s look at each element individually.
<!-- path to JBrowse refSeqs.json file -->
<refseqs>/usr/local/tomcat/tomcat7/webapps/WebApollo/jbrowse/data/refSeqs.json</refseqs>
Location where the refSeqs.json file resides, which is created from
the data generation pipeline (see the data
generation section). By default, the JBrowse data
needs to reside in
/usr/local/tomcat/tomcat7/webapps/WebApollo/jbrowse/data. If you want
the data to reside elsewhere, you’ll need to do configure your servlet
container to handle the appropriate alias to jbrowse/data.
<annotation_track_name>Annotations</annotation_track_name>
Name of the annotation track. Leave it as the default value of
Annotations.
<!-- organism being annotated -->
<organism>Pythium ultimum</organism>
Scientific name of the organism being annotated (genus and species).
We’re annotating Pythium ultimum.
<!-- CV term for the genomic sequences - should be in the form
of "CV:TERM". This applies to all sequences -->
<sequence_type>sequence:contig</sequence_type>
The type for the genomic sequences. Should be in the form of CV:term.
Our genomic sequences are of the type sequence:contig.
<!-- path to file containing canned comments XML -->
<canned_comments>/config/canned_comments.xml</canned_comments>
File that contains canned comments (predefined comments that will be available from a pull-down menu when creating comments). It’s best not to change the default option. See the canned comments section for details on configuring canned comments.
<!-- tool to be used for sequence searching. This is optional.
If this is not setup, WebApollo will not have sequence search support -->
<sequence_search_tool>
<!-- class for handling search -->
<class>org.bbop.apollo.tools.seq.search.blat.BlatCommandLine</class>
<!-- configuration for search tool -->
<config>/config/blat_config.xml</config>
</sequence_search_tool>
Here’s the configuration for sequence search tools (allows searching your genomic sequences). WebApollo does not implement any search algorithms, but instead relies on different tools and resources to handle searching (this provides much more flexible search options). This is optional. If it’s not configured, WebApollo will not have sequence search support. There are two elements that need to be configured.
<!-- class for handling search -->
<class>org.bbop.apollo.tools.seq.search.blat.BlatCommandLine</class>
Should point to the class that will handle the search request. Searching
is handled by classes that implement the
org.bbop.apollo.tools.seq.search.SequenceSearchTool interface. This
allows you to add support for your own favorite search tools (or
resources). We currently only have support for command line Blat.
<!-- configuration for search tool -->
<config>/config/blat_config.xml</config>
File that contains the configuration for the searching plugin chosen. If you’re using Blat, you should not change this. If you’re using your own plugin, you’ll want to point this to the right configuration file (which will be dependent on your plugin). See the Blat section for details on configuring WebApollo to use Blat.
Canned comments
You can configure a set of predefined comments that will be available
for users when adding comments through a dropdown menu. The
configuration is stored in
/usr/local/tomcat/tomcat7/webapps/WebApollo/config/canned_comments.xml.
Let’s take a look at the configuration file.
<?xml version="1.0" encoding="UTF-8"?>
<canned_comments>
<!-- one <comment> element per comment.
it must contain the attribute "feature_type" that defines
the type of feature this comment will apply to.
must be be in the form of "cv:cvterm" (e.g., "sequence:gene")
<comment feature_type="sequence:gene">This is a comment for sequence:gene</comment>
-->
</canned_comments>
You’ll need one <comment> element for each predefined comment. The
element needs to have a feature_type attribute in the form of
cv:cvterm that this comment applies to. Let’s make a few comments for
feature of type sequence:gene and sequence:transcript:
<comment feature_type="sequence:gene">This is a comment for a gene</comment>
<comment feature_type="sequence:gene">This is another comment for a gene</comment>
<comment feature_type="sequence:transcript">This is a comment for a transcript</comment>
Search tools
As mentioned previously, WebApollo makes use of tools for sequence searching rather than employing its own search algorithm. The only currently supported tool is command line Blat.
Blat
You’ll need to have Blat installed and a search database with your
genomic sequences available to make use of this feature. The
configuration is stored in
/usr/local/tomcat/tomcat7/webapps/WebApollo/config/blat_config.xml.
Let’s take a look at the configuration file:
<?xml version="1.0" encoding="UTF-8"?>
<!-- configuration file for setting up command line Blat support -->
<blat_config>
<!-- path to Blat binary →
<blat_bin>ENTER_PATH_TO_BLAT_BINARY</blat_bin>
<!-- path to where to put temporary data -->
<tmp_dir>ENTER_PATH_FOR_TEMPORARY_DATA</tmp_dir>
<!-- path to Blat database -->
<database>ENTER_PATH_TO_BLAT_DATABASE</database>
<!-- any Blat options (directly passed to Blat) e.g., -minMatch -->
<blat_options>ENTER_ANY_BLAT_OPTIONS</blat_options>
</blat_config>
Let’s look at each element with values filled in.
<!-- path to Blat binary -->
<blat_bin>/home/ubuntu/Software/blat/blat</blat_bin>
We need to point to the location where the Blat binary resides. We it
installed in /usr/local/bin/blat.
<!-- path to where to put temporary data -->
<tmp_dir>/data/dataHome/web_apollo/blat/tmp</tmp_dir>
We need to point to the location where to store temporary files to be
used in the Blat search. It can be set to whatever location you’d like.
We’ll store the temporary data to /data/dataHome/web_apollo/tmp.
<!-- path to Blat database -->
<database>/data/dataHome/web_apollo/blat/db/pyu.2bit</database>
We need to point to the location of the search database to be used by
Blat. We need point it to /data/dataHome/web_apollo/blat/db/pyu.2bit.
We’re using a 2bit database in this case, but you have options on how
to generate your Blat database. See the Blat documentation for more
information.
<!-- any Blat options (directly passed to Blat) e.g., -minMatch -->
<blat_options>-minScore=100 -minIdentity=60</blat_options>
Here we can configure any extra options to used by Blat. These options
are passed verbatim to the program. In this example, we’re passing the
-minScore parameter with a minimum score of 100 and the
-minIdentity parameter with a value of 60 (60% identity). See the
Blat documentation for information of all available options.
Data generation
The steps for generating data (in particular static data) are mostly
similar to JBrowse data generation steps, with
some extra steps required. Most of the scripts for data generation
reside in /usr/local/tomcat/tomcat7/webapps/WebApollo/jbrowse/bin.
Let’s go into WebApollo’s JBrowse directory.
$ cd /usr/local/tomcat/tomcat7/webapps/WebApollo/jbrowse
It will make things easier if we make sure that the scripts in the bin
directory are executable. Note that you’ll need to have root privilege
to do so.
$ sudo chmod 755 bin/*
As mentioned previously, the data resides in the data directory by
default. Let’s create a symlink to
/data/dataHome/web_apollo/jbrowse/data to be used as the data
directory. This gives a lot of flexibility in allowing your WebApollo
instance to easily point to a new data directory. Note that you’ll need
to have root privilege to do so.
$ sudo ln -sf /data/dataHome/web_apollo/jbrowse/data
Now that we have our data directory in JBrowse, we need to copy some
files into it that are specific to WebApollo’s JBrowse. We need to copy
all of the contents from ~/WebApollo/json in our data directory.
$ cp ~/WebApollo/json/* data
The first thing we need to do before processing our evidence is to
generate the reference sequence data to be used by JBrowse. We’ll use
the prepare-refseqs.pl script.
$ bin/prepare-refseqs.pl --fasta ~/maker_output/scf1117875582023.fa
We now have the DNA track setup. WebApollo’s DNA track is modified from
JBrowse’s stock DNA track, so we need to modify the newly generated
data/trackList.json file. This step IS VERY IMPORTANT, otherwise
WebApollo will not work.
First, let’s look at the stock generated data/trackList.json file.
{
"tracks" : [
{
"chunkSize" : 20000,
"urlTemplate" : "seq/{refseq}/",
"type" : "SequenceTrack",
"label" : "DNA",
"key" : "DNA"
}
],
"formatVersion" : 1
}
We need to modify the track object. We need to modify the type from
SequenceTrack to AnnotSequenceTrack, remove the urlTemplate , move
the chunkSize element into the config member we’ll be adding:
"config" : {
"chunkSize" : 20000,
"residuesUrlTemplate" : "seq/{refseq}/",
"urlTemplate" : "sequence_alterations_trackData.json",
"style" : {
"uniqueIdField" : "id",
"className" : "{type}"
},
"compress" : 0,
"type" : 1,
"subfeatures" : 1
}
After the modifications, the file should look as follow (you can just copy and paste it):
{
"tracks" : [
{
"type" : "AnnotSequenceTrack",
"label" : "DNA",
"key" : "DNA",
"config" : {
"chunkSize" : 20000,
"residuesUrlTemplate" : "seq/{refseq}/",
"urlTemplate" : "sequence_alterations_trackData.json",
"style" : {
"uniqueIdField" : "id",
"className" : "{type}"
},
"compress" : 0,
"type" : 1,
"subfeatures" : 1
}
}
],
"formatVersion" : 1
}
We need to symlink the data/seq/refSeqs.json file in data/seq.
$ cd data
$ ln -sf seq/refSeqs.json
$ cd ..
Static data generation
Generating data from GFF3 works best by having a separate GFF3 per
source type. The output from Maker puts all sources in the same GFF3. We
need to split up the GFF3. We can use the split_gff_by_source.pl
script in ~/WebApollo/tools/data to do so. We’ll output the split GFF3
to some temporary directory (we’ll use ~/scratch/split_gff).
$ mkdir -p ~/scratch/split_gff
$ ~/WebApollo/tools/data/split_gff_by_source.pl -i ~/maker_output/scf1117875582023.gff -d ~/scratch/split_gff
If we look at the contents of ~/scratch/split_gff, we can see we have
the following files:
$ ls ~/scratch/split_gff
blastn.gff est2genome.gff protein2genome.gff repeatrunner.gff
blastx.gff maker.gff repeatmasker.gff snap_masked.gff
We need to process each file and create the appropriate tracks.
We’ll start off with maker.gff. We need to handle that file a bit
differently than the rest of the files since the GFF represents the
features as gene, transcript, exons, and CDSs.
$ bin/flatfile-to-json.pl --gff ~/scratch/split_gff/maker.gff \
--arrowheadClass trellis-arrowhead --getSubfeatures \
--subfeatureClasses '{"wholeCDS": "null", "CDS":"maker-CDS", "UTR": "maker-UTR", "exon":"maker-exon"}' \
--cssClass maker-transcript --type mRNA --trackLabel maker
Now we need to convert the Maker track into WebApollo compatible JSON.
We can use json_converter.sh in ~/WebApollo/tools/data to do so.
$ ~/WebApollo/tools/data/json_converter.sh -d data/tracks/maker
root track dir: data/tracks/maker
editing files in place
seq dir count: 1
done processing features, total scanned: 353
Lastly we need to modify the maker entry in data/trackList.json.
Let’s look at the stock entry.
{
"autocomplete" : "none",
"style" : {
"className" : "maker-transcript",
"subfeatureClasses" : {
"wholeCDS" : "null",
"exon" : "maker-exon",
"CDS" : "maker-CDS",
"UTR" : "maker-UTR"
},
"arrowheadClass" : "trellis-arrowhead"
},
"key" : "maker",
"phase" : null,
"urlTemplate" : "tracks/maker/{refseq}/trackData.json",
"compress" : 0,
"label" : "maker",
"type" : "FeatureTrack",
"subfeatures" : 1
}
We need to add "renderClassName" : "maker-transcript-render" to the
style member. We also need to change type from FeatureTrack to
DraggableFeatureTrack. This is how the updated entry should look like:
{
"autocomplete" : "none",
"style" : {
"renderClassName" : "maker-transcript-render",
"className" : "maker-transcript",
"subfeatureClasses" : {
"wholeCDS" : "null",
"exon" : "maker-exon",
"CDS" : "maker-CDS",
"UTR" : "maker-UTR"
},
"arrowheadClass" : "trellis-arrowhead"
},
"key" : "maker",
"phase" : null,
"urlTemplate" : "tracks/maker/{refseq}/trackData.json",
"compress" : 0,
"label" : "maker",
"type" : "DraggableFeatureTrack",
"subfeatures" : 1
}
Now we need to process the other remaining GFF3 files. The entries in those are stored as “match/match_part”, so they can all be handled in a similar fashion.
We’ll start off with blastn as an example.
$ bin/flatfile-to-json.pl --gff ~/scratch/split_gff/blastn.gff \
--arrowheadClass trellis-arrowhead --getSubfeatures \
--subfeatureClasses '{"match_part": "blastn-alignment-part"}' \
--cssClass "blastn-alignment-match" --trackLabel blastn
Now we need to modify the blastn entry in data/trackList.json. Let’s
look at the stock entry.
{
"autocomplete" : "none",
"style" : {
"className" : "blastn-match",
"subfeatureClasses" : {
"match_part" : "blastn-part"
},
"arrowheadClass" : "trellis-arrowhead"
},
"key" : "blastn",
"phase" : null,
"urlTemplate" : "tracks/blastn/{refseq}/trackData.json",
"compress" : 0,
"label" : "blastn",
"type" : "FeatureTrack",
"subfeatures" : 1
}
We need to add "renderClassName" : "blastn-alignment-render" to the
style member. We also need to change type from FeatureTrack to
DraggableFeatureTrack. This is how the updated entry should look like:
{
"autocomplete" : "none",
"style" : {
"renderClassName" : "blastn-alignment-render",
"className" : "blastn-match",
"subfeatureClasses" : {
"match_part" : "blastn-part"
},
"arrowheadClass" : "trellis-arrowhead"
},
"key" : "blastn",
"phase" : null,
"urlTemplate" : "tracks/blastn/{refseq}/trackData.json",
"compress" : 0,
"label" : "blastn",
"type" : "DraggableFeatureTrack",
"subfeatures" : 1
}
We need to follow the same steps for the remaining GFF3 files. It can be
a bit tedious to do this for the remaining six files, so we can use a
simple inline shell script to help us out. However, we’ll still have to
manually modify data/trackList.json for each new track. Don’t worry if
the script doesn’t make sense, you can still process each file by hand.
For now you can just copy and paste the script into your terminal.
$ for i in $(ls ~/scratch/split_gff/*.gff | grep -v maker);
do j=$(basename $i); j=${j/.gff/};
echo "Processing $j" && bin/flatfile-to-json.pl --gff $i --arrowheadClass trellis-arrowhead \
--getSubfeatures --subfeatureClasses "{\"match_part\": \"$j-alignment-part\"}" \
--cssClass "$j-alignment-match" --trackLabel $j; done
IMPORTANT!
Remember that we still need to add
"renderClassName" : "$type-alignment-render" to the style member,
where $type is the track name. We also need to change type from
FeatureTrack to DraggableFeatureTrack.
Now let’s look how to configure BAM support. WebApollo has native support for BAM, so extra processing of the data is required.
First we’ll copy the BAM data into the WebApollo data directory. We’ll
put it in the data/bam directory. Keep in mind that this BAM data was
randomly generated, so there’s really no biological meaning to it. We
only created it to show BAM support.
$ mkdir data/bam
$ cp ~/maker_output/*.bam* data/bam
Now we need to edit the data/bam_trackList.json file. Let’s look at
the file.
{
"tracks" : [
{
"type" : "BamFeatureTrack",
"label" : "ENTER_LABEL",
"data_url" : "ENTER_URL_TO_BAM_FILE",
"index_url" : "ENTER_URL_TO_BAI_FILE",
"config" : {
"autocomplete" : "none",
"style" : {
"className" : "bam",
"subfeatureClasses" : {
"M": "cigarM",
"D": "cigarD",
"N": "cigarN",
"=": "cigarEQ",
"E": "cigarEQ",
"X": "cigarX",
"I": "cigarI"
},
"arrowheadClass" : null
},
"key" : "ENTER_KEY",
"compress" : 0,
"type" : 1,
"subfeatures" : 1
}
}
],
"formatVersion" : 1
}
We need to modify the following elements: label, data_url,
index_url, and key.
Let’s look at each value with their values filled in.
"label" : "simulated_bam",
The internal name for this BAM track.
"data_url" : "bam/scf1117875582023.bam",
URL to BAM file.
"index_url" : "bam/scf1117875582023.bam.bai",
URL to BAM index file.
"sourceUrl" : "data/",
Add sourceUrl under index_url.
You can point to the BAM/BAI file in any server by providing an absolute URL. This is a nice feature since BAM files can get rather large, so you don’t need to create a local copy on your own server.
"key" : "simulated BAM",
The external, human-readable label seen on the BAM track.
Here’s how the updated JSON should look like.
{
"tracks" : [
{
"type" : "BamFeatureTrack",
"label" : "simulated_bam",
"data_url" : "bam/scf1117875582023.bam",
"index_url" : "bam/scf1117875582023.bam.bai",
"sourceUrl" : "data/",
"config" : {
"autocomplete" : "none",
"style" : {
"className" : "bam",
"subfeatureClasses" : {
"M": "cigarM",
"D": "cigarD",
"N": "cigarN",
"=": "cigarEQ",
"E": "cigarEQ",
"X": "cigarX",
"I": "cigarI"
},
"arrowheadClass" : null
},
"key" : "simulated BAM",
"compress" : 0,
"type" : 1,
"subfeatures" : 1
}
}
],
"formatVersion" : 1
}
You should now have a simulated BAM track now available.
You can download the updated trackList.json file here (you’ll need to
put the file in your virtual machine and unzip it in
/usr/local/tomcat/tomcat7/webapps/WebApollo/jbrowse/data):
web_apollo_track_list_json.zip
Alternatively, you can just copy the WebApolloDemo trackList.json into
WebApollo (since they’re both using the same dataset).
$ cd /usr/local/tomcat/tomcat7/webapps/WebApollo/jbrowse/data
$ cp /usr/local/tomcat/tomcat7/webapps/WebApolloDemo/jbrowse/data/trackList.json .
Congratulations, you’re done configuring WebApollo.
Let’s test out our installation. Point your browser to
http://ec2-##-##-##-##.compute-1.amazonaws.com:8080/WebApollo
(e.g.,
http://ec2-184-73-92-243.compute-1.amazonaws.com:8080/WebApollo).
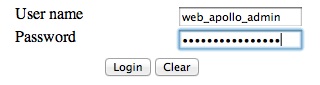
The user name and password are both web_apollo_admin as we configured
earlier.
![]()
Click on the Edit annotations button.
![]()
We only see one reference sequence to annotate since we’re only working
with one contig. Click on the Edit button.
Now have fun annotating!!!
Facts about “WebApollo Tutorial 2012”
| Has topic | WebApollo + |