Two kinds of example: the standalone react-msaview viewer, where the biology reads straight off the alignment and tree, and react-msaview running inside JBrowse, connected to a genome view and 3D structure. Each is built reproducibly from public data by the scripts in the repo. Build your own from any gene on the JBrowse integration page.
Standalone react-msaview
Just an alignment, a tree, and optional feature overlays — no genome browser. Each figure below shows a distinct viewer capability — domain overlays, reference diffing, tree topology, RNA secondary structure, gene arrows, base-resolution DNA, scale. The full set of biological examples (more conservation, phylogeny, and domain stories) is live and interactive on the examples page.
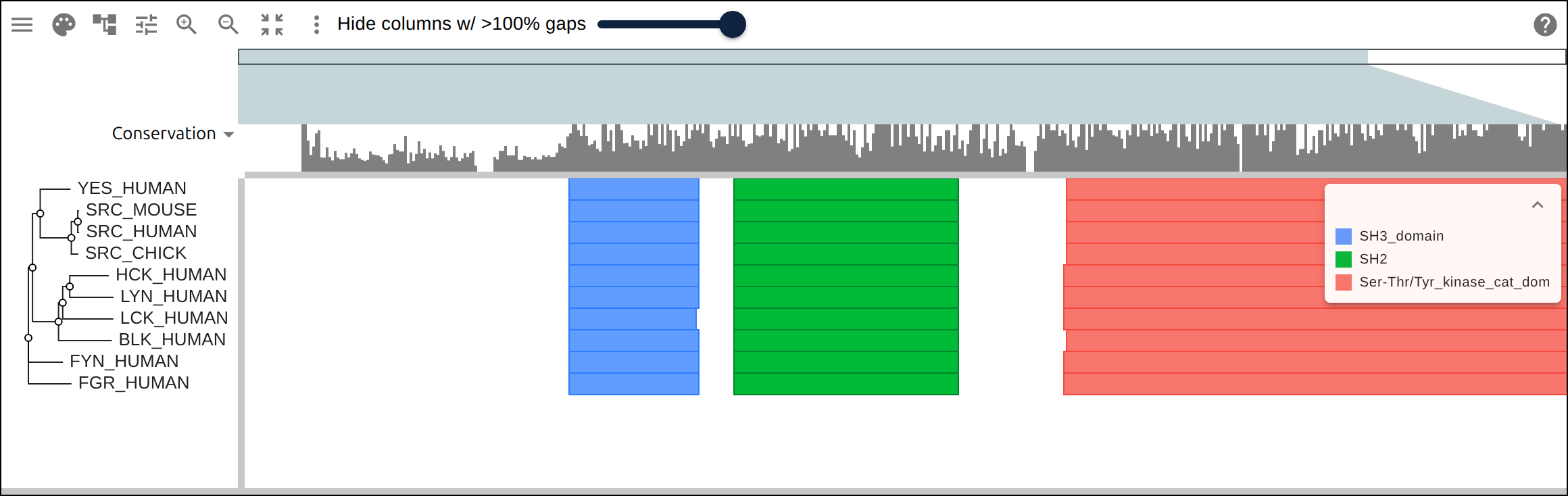
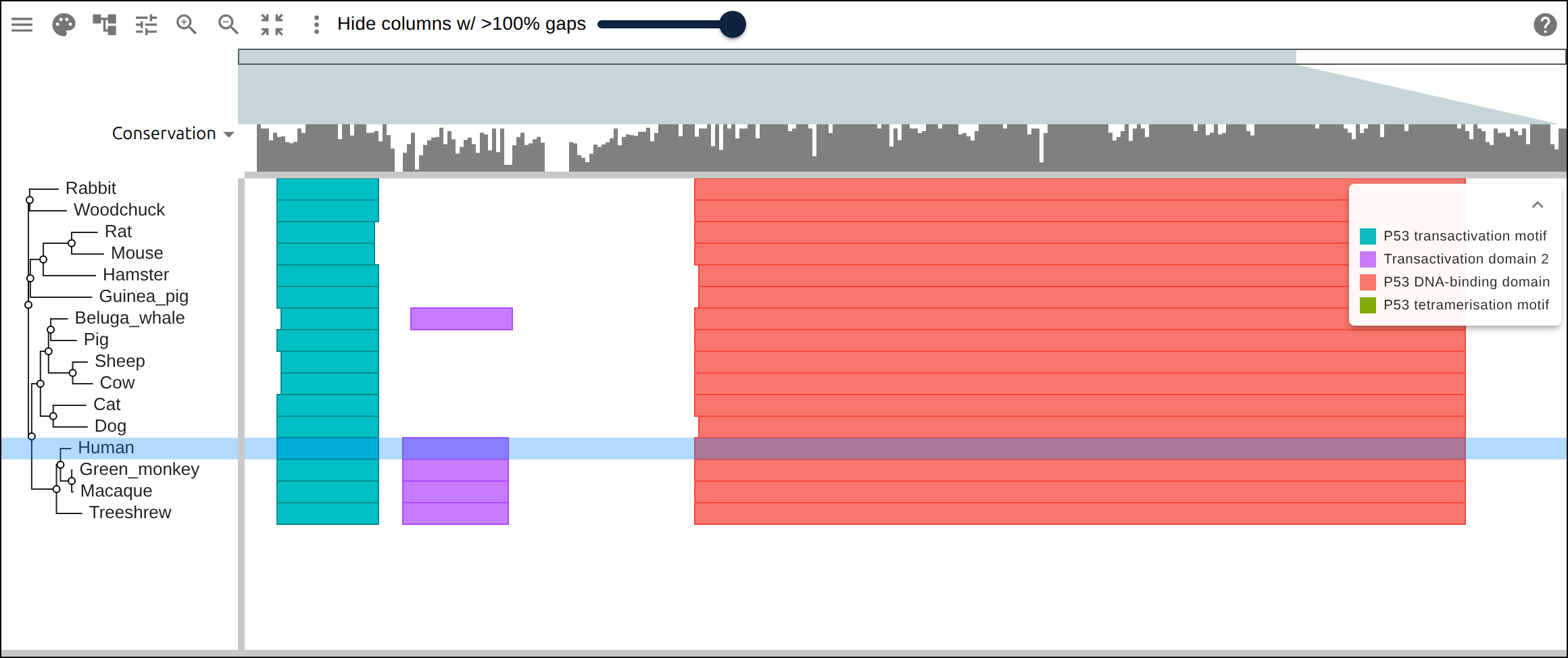
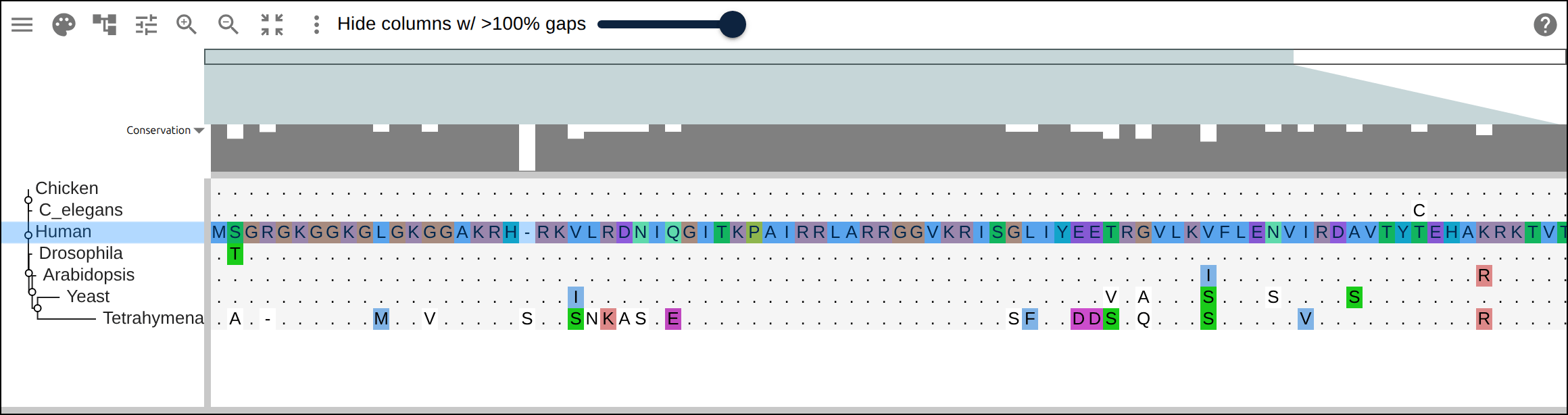
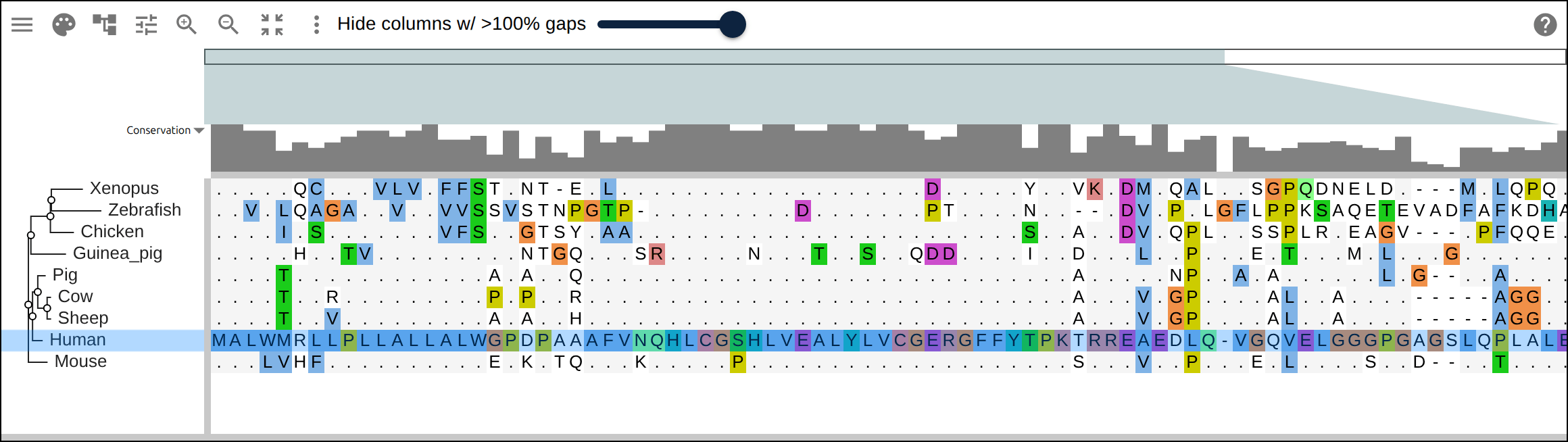
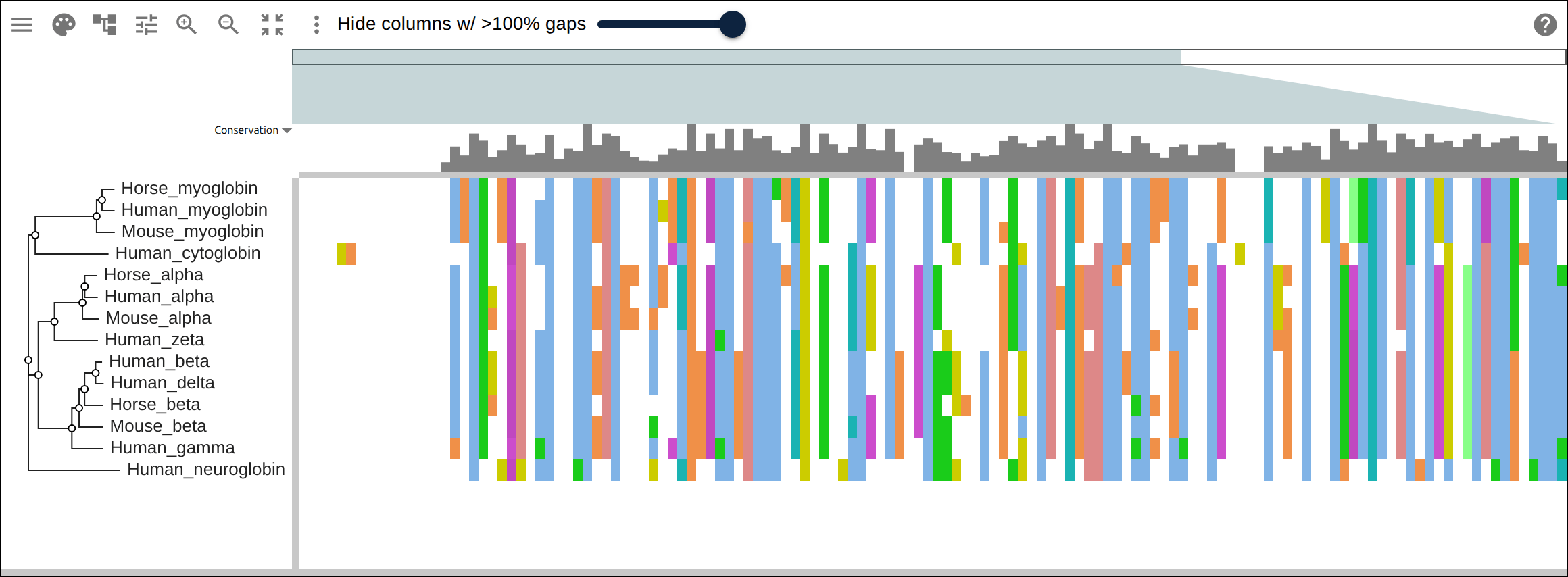
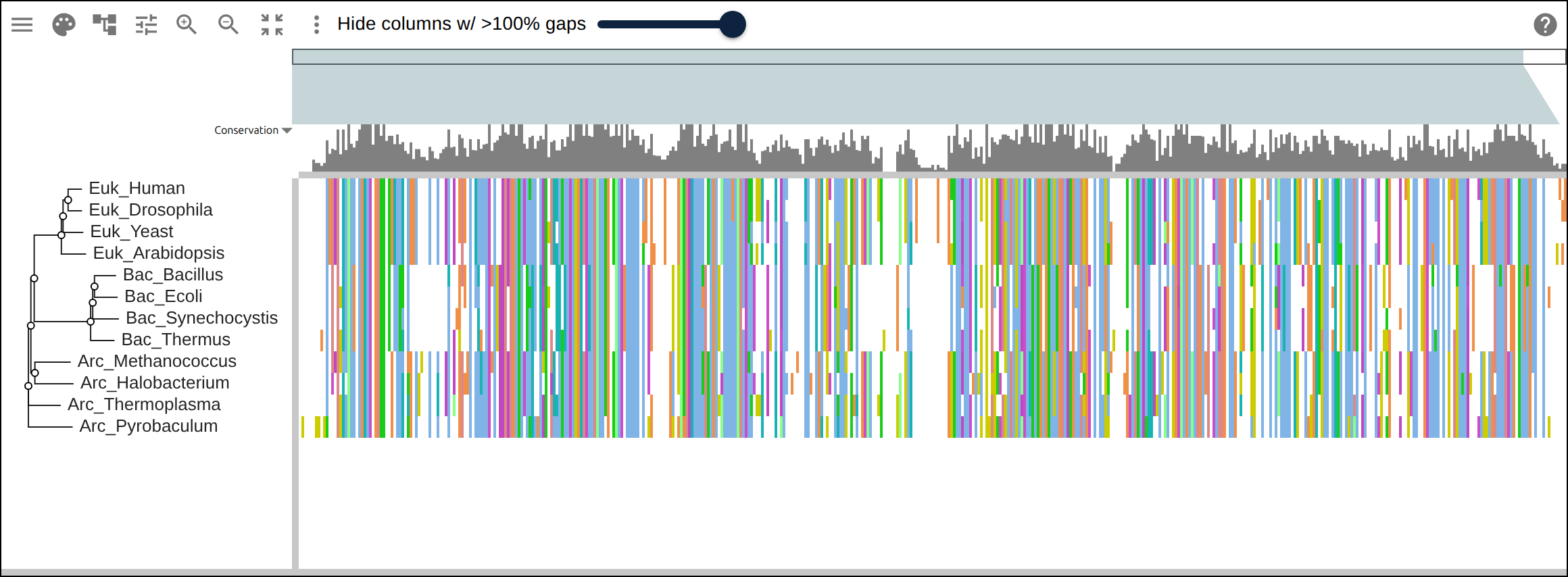
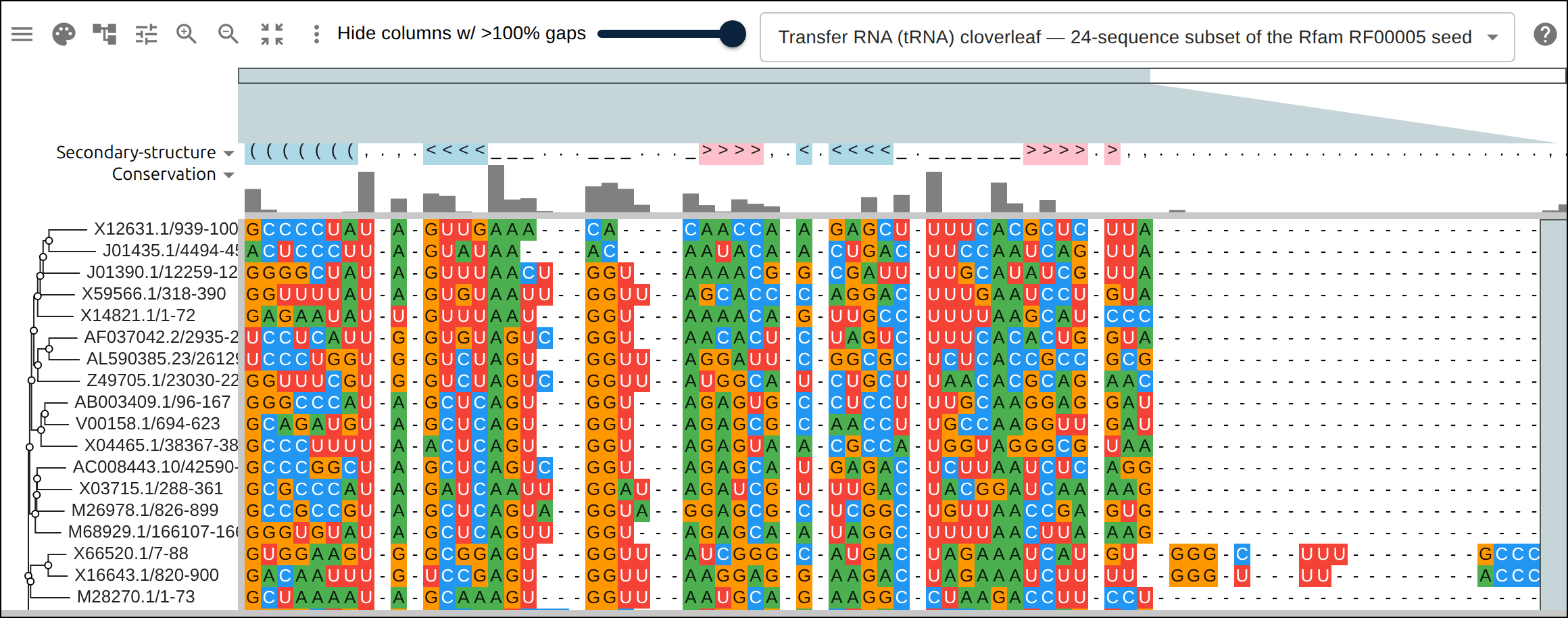
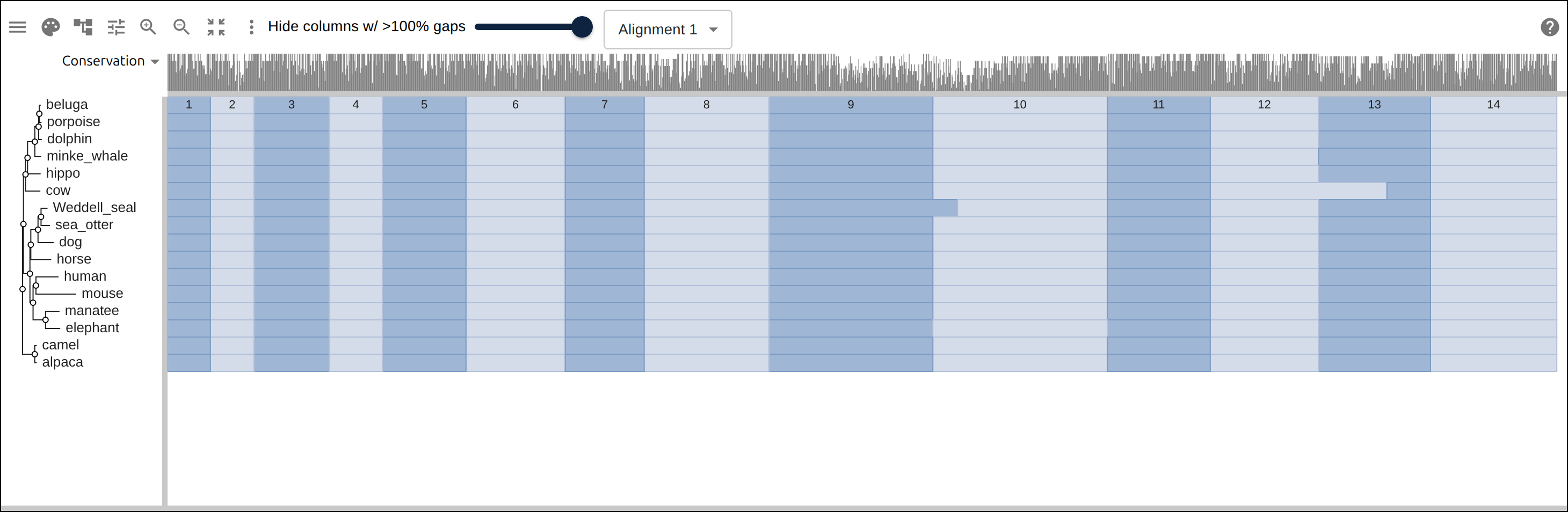
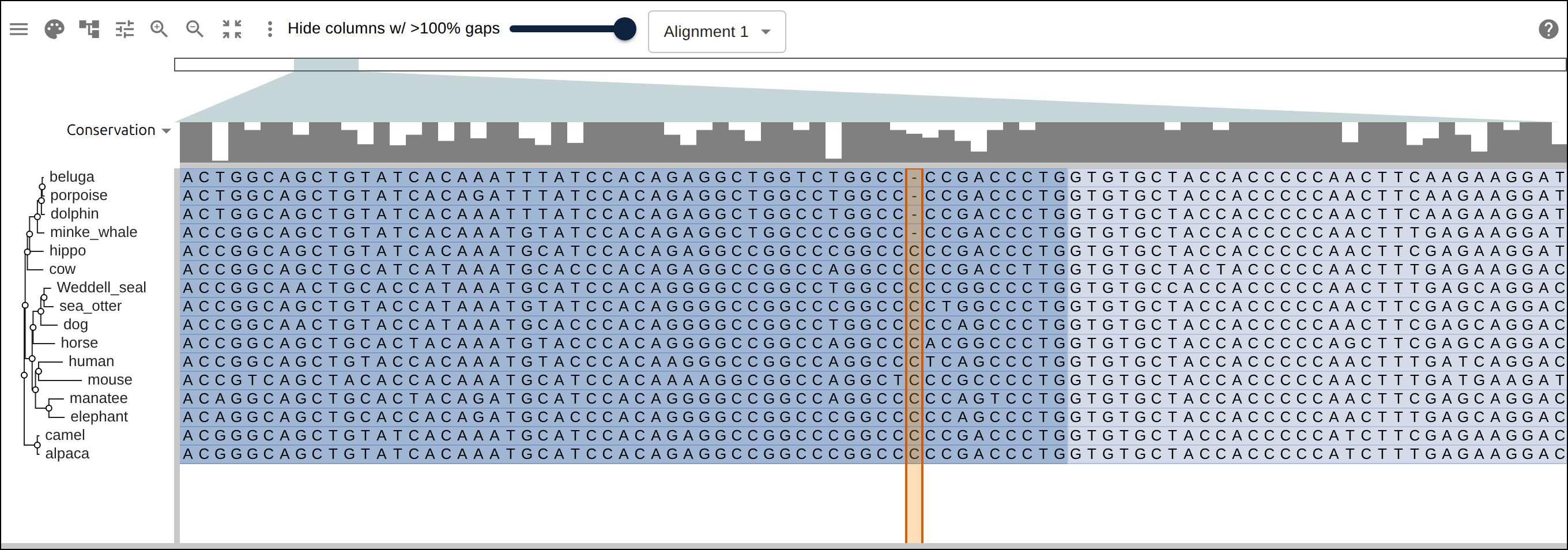
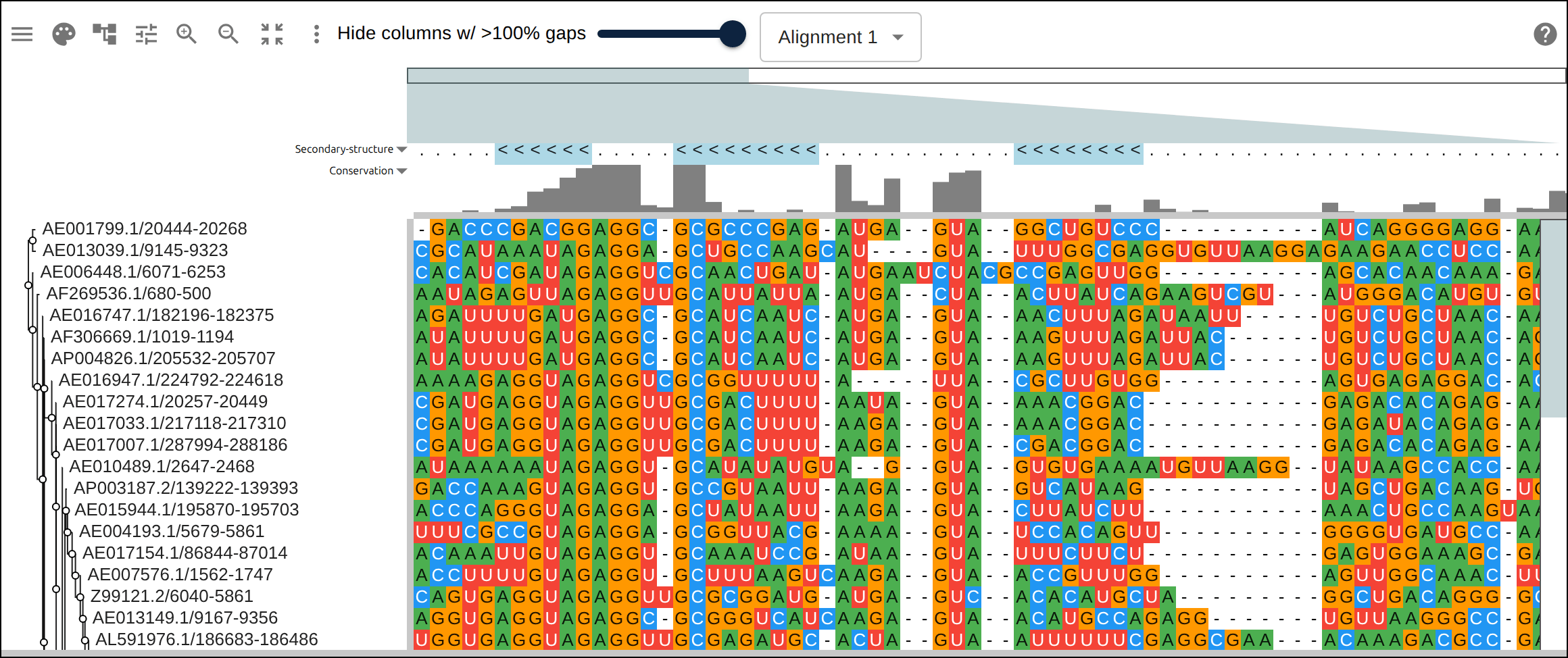
Domain architecture — The Src-family kinases with InterProScan annotations: the shared SH3 + SH2 + kinase domain blocks line up down every member, so the family’s domain layout reads straight across the alignment.A dominant functional domain — p53’s InterProScan domains overlaid: the central DNA-binding domain (red) — where most cancer mutations cluster — forms the bulk of the protein, flanked by the short N-terminal transactivation motifs. The overlay maps the functional architecture onto the alignment.Reference comparison (dots) — Diffing against a reference (relativeTo) collapses identical residues to dots so only the changes stand out. Histone H4, one of the most conserved proteins known, is almost entirely dots relative to human — only the most distant lineages differ.Post-translational processing — Insulin relative to human: the mature B and A chains stay conserved (dots) while the cleaved-out C-peptide drifts (letters) — the same dot-diffing reading out where a protein is under selection.Gene duplication — The globin family groups by globin type across species rather than by species — alpha beside alpha, beta beside beta — the signature of ancient gene duplication read off the tree.Tree of life — Elongation factor EF-1α/EF-Tu across bacteria, archaea, and eukaryotes — the label prefixes make the three domains of life read straight off a single inferred tree.RNA secondary structure — A tRNA alignment (Rfam RF00005): the Stockholm SS_cons cloverleaf renders as a dedicated secondary-structure track above the columns, with the acceptor stem and D-/anticodon-/T-arms colored by base-pairing.Gene structure overlay (DNA) — Coagulation factor XII coding alignment with its 14 exons overlaid — each exon the same color across species, through the same overlay path as InterProScan domains (built by react-msaview-cli genestructure). The whole gene’s exon architecture reads straight down the alignment.Pseudogenization (base resolution) — Zoomed to single nucleotides: F12 is intact in land mammals and the manatee, but a single-base deletion shared by exactly the four cetaceans — beside premature stops elsewhere — disables it. Gene loss visible at the base level, which a protein alignment cannot show.Gene-arrow map — A gggenes-style gene cluster across genomes, each gene a strand-directed arrow anchored to alignment columns: one gene is inverted (the arrow flips) and one deleted (its columns gap out), yet every gene stays column-aligned.Scale — A real ~60-sequence lysine-riboswitch family and its full inferred tree, rendered through the tiled canvas system that keeps large alignments responsive.
Inside JBrowse
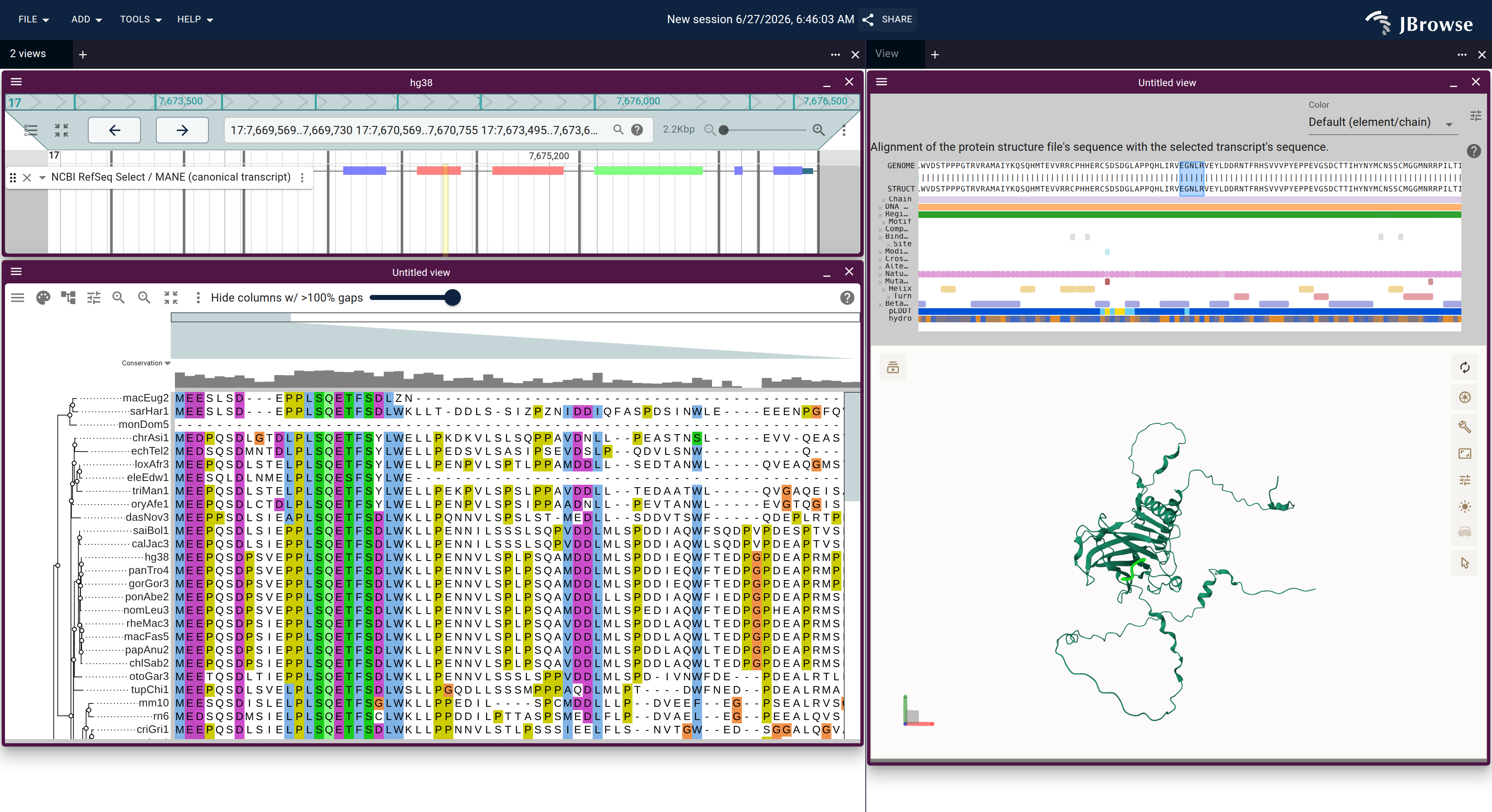
react-msaview also runs as a JBrowse 2 plugin: the alignment and tree open inside JBrowse, connected to a genome view and — with protein3d — the AlphaFold structure. Selecting a region in any view lights the matching columns, codons, and residues in the others. Each example below is one declarative URL.
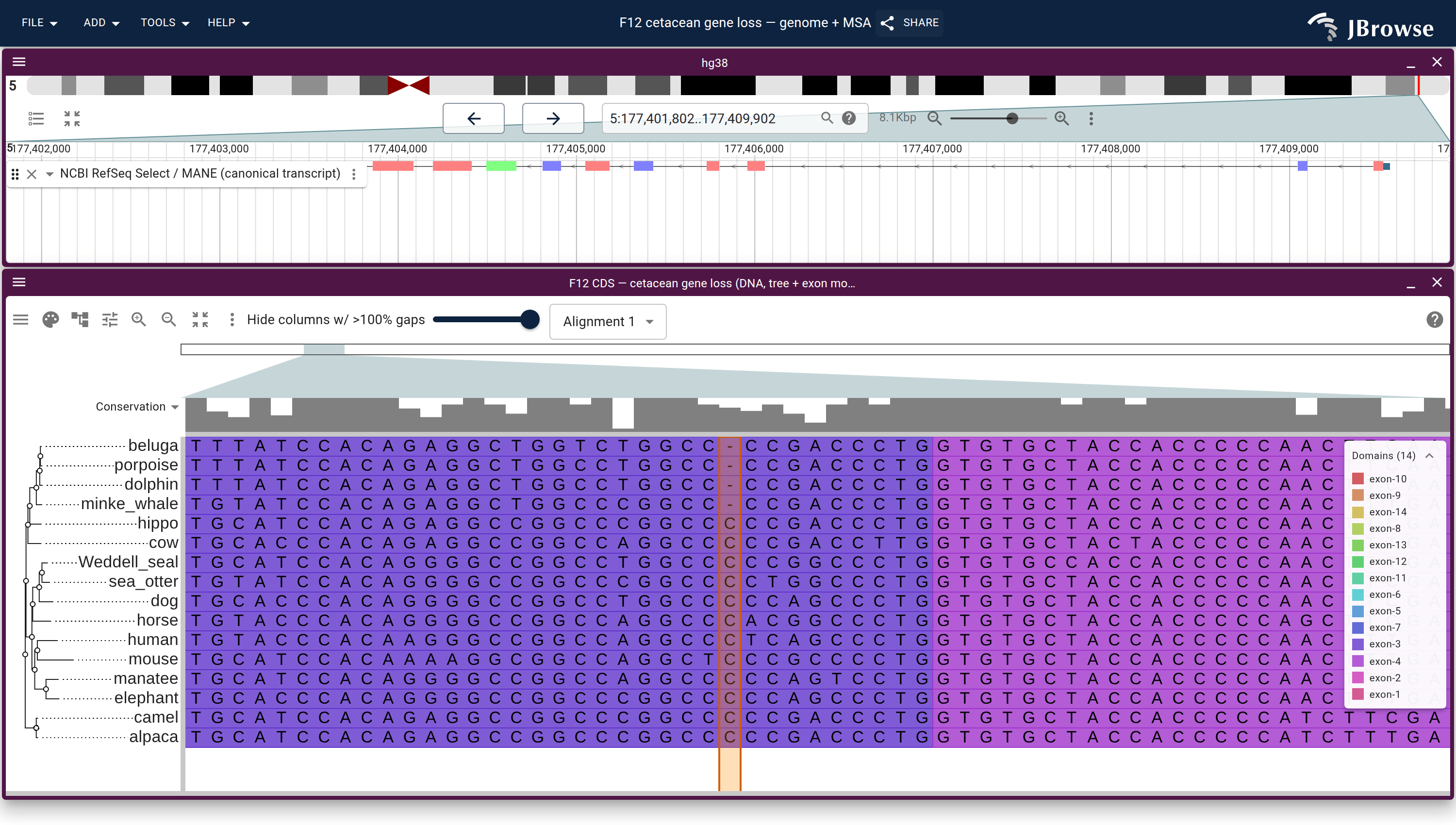
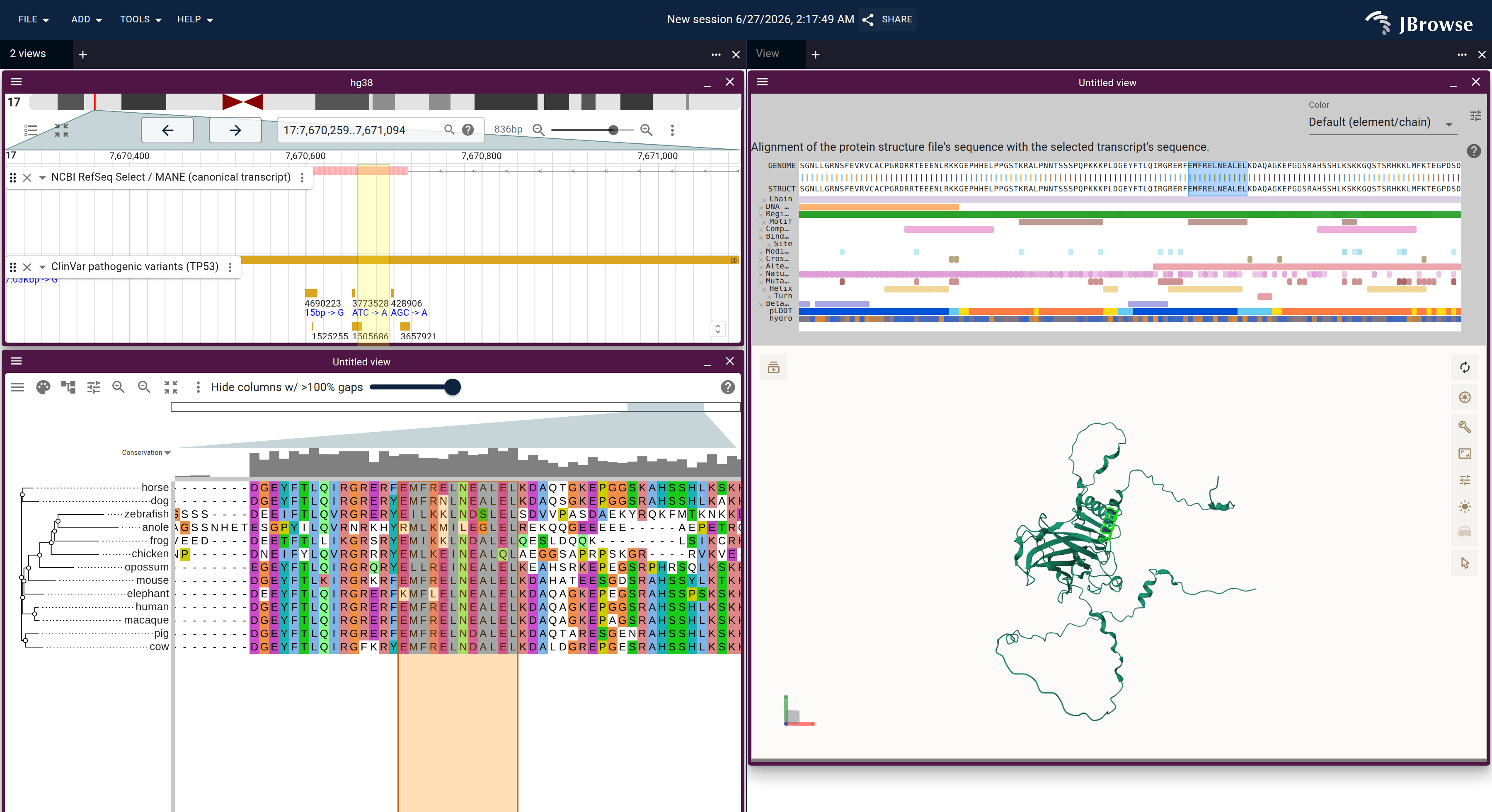
Genome ↔ alignment in one JBrowse session: the F12 locus (RefSeq annotation, top) over the react-msaview DNA alignment + species tree, zoomed to base resolution at exon 3. The highlighted column ties the genome to the alignment — the four cetaceans carry a single-base deletion (a gap where every other mammal has a C) that disables the gene. This is the viewer an MSA editor would build on: the 14-exon model is already registered to the alignment columns and lifted onto each species. Open in JBrowse ↗Genome ↔ alignment ↔ 3D structure: the TP53 gene (RefSeq + ClinVar), the p53 ortholog alignment, and the AlphaFold structure as one connected session, opened with the nuclear export signal motif (residues 339–350) lit across all three — the same motif highlighted on the genome, the alignment column, and the folded structure at once. Open in JBrowse ↗Built on the fly by the gene explorer for any human gene: selecting a span of residues on the AlphaFold structure lights the exact same codons on the collapsed-intron genome view (the yellow band) and the 100-way alignment — structure ↔ genome ↔ alignment, codon-precise.
The same connected pattern without the structure view:
SRC ↗ — the Src-family kinase alignment beside the SRC gene.
BRAF V600E ↗ — the RAF family on the invariant V600 codon, with a ClinVar pathogenic-variant track.
TP53 R248 ↗ — p53 across vertebrates on the R248 hotspot, with ClinVar.