GMOD
Using the topoview Glyph
This glyph is derived from the FlyBase fb_shmiggle Glyph, 2009-2010 Victor Strelets, FlyBase.org
It is maintained by Sheldon McKay <sheldon.mckay@gmail.com>
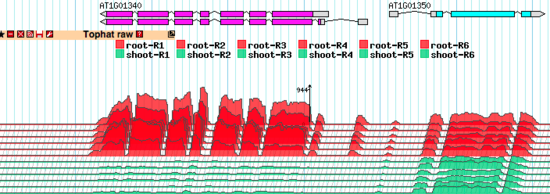
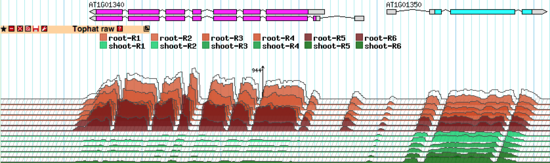
The TopoView glyph was developed for fast 3D-like demonstration of RNA-seq data consisting of multiple individual subsets. The main purposes were to compact presentation as much as possible (in one reasonably sized track) and to allow easy visual detection of coordinated behavior of the expression profiles of different subsets. See the note below about normalizing the expression profiles across the whole experiment.
Contents
Demonstration
Setting up this Glyph can be complicated. If you have access to Amazon Web Services, there is an AMI with a demonstration instance of Gbrowse using the topoview track for RNASeq data at:
GBrowse 2.56 with topoview track - ami-c2d677bf
Data format
Comparing performance (retrieval of several Kbp of data profiles for several subsets of some RNA-seq experiment) of wiggle binary method and of several possible alternatives, it was discovered that one of the approaches remarkably outperforms wiggle bin method (although it requires several times more space for formatted data storage). Optimal storage/retrieval method stores all experiment data (all subsets of the experiment) in one text file, where structure of the file in fact is one of the most simple wiggle (coverage files) formats with the addition of some positioning data (two-column format, without runlength specification, without omission of zero values). This is the only format which glyph is able to handle.
# subset =BS107_all_unique chromosome =2LHet
-200000 0
0 0
19955 1
19959 0
19967 2
19972 0
19977 2
20027 0
20031 2
20035 0
20043 1
Accessory scripts
The Bio::Graphics package has two scripts useful for processing BAM alignment data from programs such as tophat for use with this glyph.
Converting BAM alignment files to WIG coverage files
bam_coverage_windows.pl accepts a sorted bam file as input and will calculate the average read coverage for a user-specified window size (default 25). The windows are non-overlapping. The output format is WIG/BED4, which is the format used by the coverage_to_topoview.pl script.
Usage: bam_coverage_windows.pl -b bamfile -n 10_000_000 -w 25 | gzip -c > bamfile.wig.gz
-b name of bam file to read REQUIRED
-w window size (default 25)
-n normalized read number -- if you will be comparing multiple bam files
select the read number to normalize against.
All counts will be adjusted by a factor of:
actual read count/normalized read count
The output of this script looks like (note the zero-based, half-open coordinates):
track type=wiggle_0 name="root-R1" description="read coverage for bam/root-R1.bam (window size 25)"
1 3625 3650 0.7617701464
1 3650 3675 9.3417075848
1 3675 3700 18.5230425072
1 3700 3725 26.5817687928
1 3725 3750 35.682917384
1 3750 3775 40.494097256
1 3775 3800 45.0246249688
1 3800 3825 52.2413947768
NOTE: Normalization
If you are comparing BAM files with different total read counts, you need to normalize the read counts across BAM file with the -n option. The number used is arbitrary. Try using a number near the average read count for all BAM files being analyzed in the experiment.
Converting coverage WIG to indexed data for topoview
coverage_to_topoview.pl converts a list of coverage files (WIG/BED4) to the indexed format used by this glyph. It specifically expects the BED4 format produced by the bam_coverage_windows.pl script
Usage: coverage_to_topoview.pl [-o output_dir] [-h] [-l] file1.wig.gz file2.wig.gz
-o output directory (default 'topoview')
-l use log2 for read counts (recommended)
-h this help message
Log transformations
The dynamic range in read coverage can be very high and log2 conversion dramatically changes perception of expression profiles and helps to illuminate coordinated behavior of different subsets. Using log2-transformed read counts in addition to raw read counts is recommended.
Subsets (subplots)
Each WIG file corresponds to a single BAM file and will be a subset in the experiment. The base name of the file will be used as the subset name. For example, the file shoots-R1.wig.gz will generate a subset named shoots-R1 in the topoview track.
Output directories
If you do not specify an output directory name, the default name ‘topoview’ will be used. Any existing contents will be overwritten. For example, if you are making two tracks, one using raw counts and the other using log2 transformed counts:
coverage_to_topoview.pl -o raw file1.wig.gz file2.wig.gz
coverage_to_topoview.pl -o log2 -l file1.wig.gz file2.wig.gz
..will yield:
├── log2
│ ├── data.cat
│ └── index.bdbhash
└── raw
├── data.cat
└── index.bdbhash
The absolute path of these directories will be used in the datadir option below.
Configuration
Example config stanza
[TOPOVIEWLOG2]
feature = region
glyph = topoview
autoscale = local
height = 200
datadir = /home/ubuntu/data/bam/log2
subset order = SRR1810778.25 FF9966
SRR1810779.25 FF6633
SRR1810780.25 FF0000
SRR1810781.25 00CC66
SRR1810782.25 009933
SRR1810783.25 006600
key = TopHat: Normalized Read Coverage (log2)
show max = 0
x_step = 2
y_step = 8
fill opacity = 0.8
edge color = black
Options
Glyph-specific options
- feature: The full-length feature for the track. This would usually be the feature type you configured for your chromosomes or scaffolds. You don’t need to add more GFF data for this track
- database: The same database as you used for the chromosomes
- autoscale: options are ‘local’ and ‘global’. local scales to the on-screen max value, global scales to the global max
- datadir: location of the indexed coverage data (absolute path)
- show max: show an extra subset corresponding to the maximum coverage across all subsets
- x_step: the horizontal offset (pixels) of each plotted subset (can not be zero)
- y_step: the vertical offset (pixels) of each plotted subset (can not be zero)
- fill opacity: the degree of transparency of each plotted subset. Translucency aids in the comparison.
- edge color: the outline color for the plots (default: charcoal gray)
- subset order the order and color of each subset (subplot) in the graph (see below)
Subsets
Setting the ‘subset order’ is mandatory. It specifies the subplots and the order in which they will be displayed.
There are three ways to represent the subsets:
Ordered subsets with no color specified. Random colors will be assigned. Hope you are feeling lucky.
subset order = SRR1810778.25
...
Ordered subsets with color specified (use either web colors or hex colors with the ‘#’ omitted)
subset order = SRR1810778.25 red
SRR1810779.25 FF6633
...
Ordered subsets with color and opacity set. Not that the global ‘fill opacity’ option affects all subsets. Specifying individual opacity is optional.
subset order = SRR1810778.25 FF9966 0.8
SRR1810779.25 FF6633 0.7
...
Installation Troubleshooting
Track has a red error message
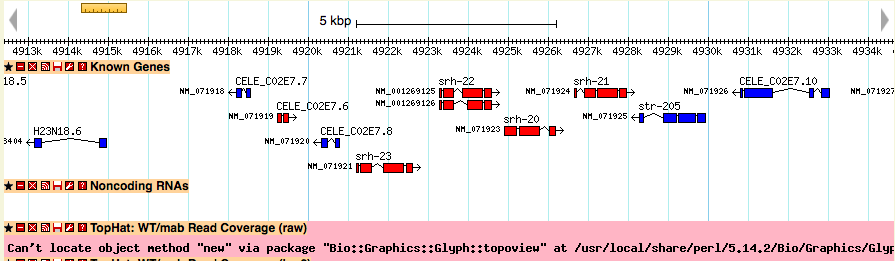
The glyph uses the Perl interface to BerkeleyDB. If you see a red error message in where the track should be, this might be why. In an Ubuntu/debian system, try:
sudo apt-get install libdb5.3 libdb5.3-dev libberkeleydb-perl
You may need to use apt-cache search to find other version of the above Debian packages.
- For redhat based systems, such as centOS, use yum to search for equivalent packages.
Track is present but has no data

Make sure the “feature=” option is set to the same kind of kind of feature as the chromosomes in your scaffolds database
For example: If you were using the chromosome GFF below, you would set your topoview feature to ‘region’:
##gff-version 3
V ce10 region 1 20924149 . . . ID=V;Name=V
X ce10 region 1 17718866 . . . ID=X;Name=X
IV ce10 region 1 17493793 . . . ID=IV;Name=IV
II ce10 region 1 15279345 . . . ID=II;Name=II
I ce10 region 1 15072423 . . . ID=I;Name=I
III ce10 region 1 13783700 . . . ID=III;Name=III
M ce10 region 1 13794 . . . ID=M;Name=M
Some other kind of error
Make sure your software is up-to-date
- Bio::Graphics version 2.4 or greater
- BioPerl 1.7.0 or greater
- GBrowse 2.54 or greater
Ask for help
sheldon.mckay@gmail.com