GMOD
Arthropod Genomics 2011/Genome Project 101 Workshop
(Redirected from AGS2011
 |
Genome Project 101 Workshop Arthropod Genomics Symposium
2011 |
This is the wiki page for the Genome Project 101 Workshop presented at Arthropod Genomics 2011.
This page serves as the slides for the workshop. It will be tidied up within a week of the end of the workshop.
Contents
The One True URL
Everything you need to know from this workshop can be found on this page. The short URL is:
VMware Image
A VMware image was used during this workshop. That image can be downloaded, installed, and run locally on your system. See VMware for details.
Worked Examples
MAKER Web Annotation Service
While we could install MAKER locally on this machine, it is nice to be able to make use of the web service provided by Mark Yandell’s group at University of Utah. To use it, go to
http://derringer.genetics.utah.edu/cgi-bin/MWAS/maker.cgi
and create a free account (I created one for this tutorial with a user name of gmodags). After that is created, we can upload some sample data. I put the sample data that I used on ~/Downloads/MAKER_input, where there are three files:
- pyu-contig.fasta - a FASTA file containing a 1.7 MB contig
- pyu-est.fasta - A set of assembled 454 read ESTs from P. ultimum and a related organism
- pyu-protein.fasta - a set of protein sequence from a related organism
After clicking on the “New Job” tab, I uploaded all three files in the appropriate spot, ignoring the others:
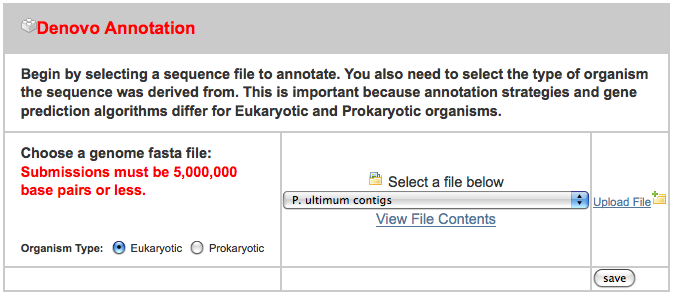
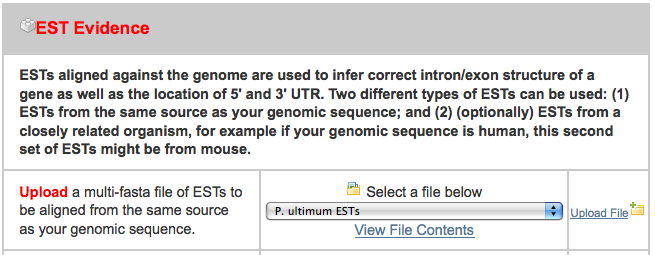
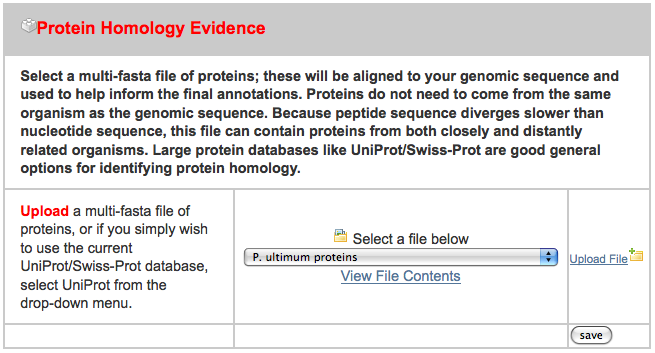
After uploading these, I pressed “Add to Job Queue” to get it started running. The job waited under an hour before starting, and then finished in under three hours.
Upon finishing, I was presented with multiple ways of looking at the data:
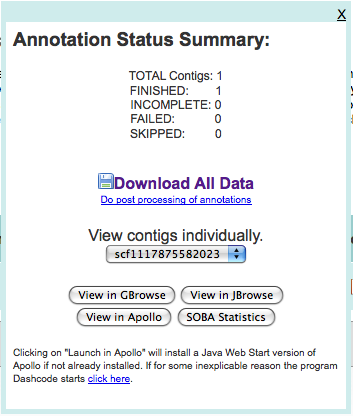
and after taking a quick look at both GBrowse and JBrowse, downloaded the data to the machine (in ~/Downloads/3263.maker.output). The GFF file in this directory will be loaded into Chado.
Galaxy Example
We have our annotation, let’s do some further analysis and exploration.
To further analyze your data you need tools like Galaxy, BioMart and InterMine. Since I work for Galaxy, we’ll spend some time working on a simple example in that. We’ll touch on BioMart or InterMine as time allows.
1. Get to Galaxy
We could run this analysis on the free public Galaxy server (http://usegalaxy.org), or on the Galaxy that has been installed on our VMware image. Let’s run it on our local install.
Note: Please don’t run on the local install with me. The public server might be able to support 120 people doing this simultaneously. The local install won’t. Galaxy
2. What have we got?
First load the GFF that MAKER produced into Galaxy
Get Data → Upload File → ftp://ftp.gmod.org/pub/gmod/Meetings/2011/AGS/3263.maker.output/3263.all.gff → Execute
This uploads the GFF file into Galaxy. It recognizes it as a GFF3 file.
Now, because of a bug in Galaxy (don’t tell anyone), we need to convert it to BED to run a subsequent step.
Convert Formats → GFF-to-BED → Execute
Now lets see what is in the annotation. Lets count the number of different feature types in the file.
Join, Subtract and Group → Group → Group by Column: c4
This tells Galaxy please group the lines by the value in column 4, which is the SO type of the feature
Add new operation → Type: Count → Execute
Now count the number of lines that have each type.
Anything interesting? Hmmm. We’ve got one more exon than CDS. I wonder where that is?
3. Get just the Exons and CDSs
Just get the exons:
Filter and Sort → Filter → Filter: GFF-to-Bed on data
The SO type is in column 4 in BED.
With following condition: c4==’exon’ → Execute
Repeat with CDS.
4. See what is in the exon set that is not in the CDS set
Operate on Genomic Intervals → Subtract
Subtract CDSs from Exons → Execute
5. Investigate
We have one exon left. Go visualize it in GBrowse or JBrowse.
For the long term
This history has been run and saved on the public Galaxy instance. Go to Shared Data → Published Histories → AGS2011. It has also been saved on the Galaxy instance on the VMware image under user gmodags (password same as user).
Chado
GBrowse
We can go to http://localhost/cgi-bin/gb2/gbrowse/pythium (or http://gmodags.bx.psu.edu/cgi-bin/gb2/gbrowse/pythium during the workshop).
JBrowse
Go to http://localhost/jbrowse (or http://gmodags.bx.psu.edu/jbrowse during the workshop).
VMware Image
A VMware image will be made available to participants of the workshop. We will use this image during the workshop
System Configuration
This section attempts to track what we did to create the VMware image
| Operating System | Ubuntu 11.04, 64 bit client. This is a popular Linux distribution |
| Memory | 2 GB. If you run this on a system that has 2 gigabytes or less of memory, please decrease this number |
| Disk | 80 GB. This is allocated 2 GB at a time, as needed, but VMware. |
| Networking | NAT |
| Username | gmod |
| Password |
Installed Prerequisite Software
GMOD components have a variety of prerequisite software that needs to be installed. Here is a list of what was installed so we could install and run GMOD software.
| Software | How | Comments |
|---|---|---|
| Mercurial | sudo apt-get install mercurial |
Revision control system used by Galaxy |
| Microsoft TrueType core fonts | sudo apt-get install ttf-mscorefonts-installer |
Used by Galaxy. |
| python-dev | sudo apt-get install python-dev |
Used in Galaxy. |
| python-setuptools | sudo apt-get install python-setuptools |
Used in Galaxy. |
| python-pip | sudo apt-get install python-pip |
Used in Galaxy. |
| bx-python scripts | sudo pip install bx-python |
Scripts used by Galaxy |
| Python 2.6 | sudo apt-get install python2.6 |
Ubuntu 11.04 comes with Python 2.7, which Galaxy, does not like. This installs 2.6 in parallel. |
| Python R package interface | sudo apt-get install python-rpy |
Used in basic Galaxy operations. |
| Graphics libraries | sudo apt-get install libgd2-xpm-dev libgd-gd2-perl libgd-tools libgd-svg-perl |
Used by GBrowse |
| System utilities and web server | sudo apt-get install autoconf apache2 |
Used by GBrowse and Chado |
| Database server | sudo apt-get install postgresql postgresql-client |
Used by Chado and GBrowse |
| Variety of perl modules | sudo apt-get install libcgi-session-perl libdbd-pg-perl libdigest-md5-file-perl libclass-base-perl libmodule-build-perl libstatistics-descriptive-perl libtemplate-perl libxml-simple-perl liblog-log4perl-perl libparse-recdescent-perl libsql-translator-perl perl-doc |
Used by Chado and GBrowse |
| Perl modules | sudo apt-get install libjson-xs-perl libdevel-size-perl |
Used by JBrowse |
| Perl graphics library | cpan> install GD |
Used by JBrowse and GBrowse |
| BioPerl libraries | cpan> install Bio::Perl Bio::Graphics JSON |
Used by JBrowse, GBrowse and Chado |
| GBrowse Chado adaptor | cpan> install Bio::DB::Das::Chado |
Used by JBrowse, GBrowse and Chado |
| More perl libraries | cpan> install GO::Parser Module::Load DBIx::DBSchema XML::Parser::PerlSAX |
Used by Chado |
| Even more perl libraries | cpan> install Heap::Simple Heap::Simple::Perl Heap::Simple::XS PerlIO::gzip |
Used by JBrowse |
PostgreSQL Configuration
The postgresql server will be set up with fairly unrestricted access to make life easier during the tutorial. If used “in real life”, the configuration should be tightened down quite a bit.
Edit config file
sudo su -
vi /etc/postgresql/8.4/main/pg_hba.conf
Change the bottom lines to look like this:
# "local" is for Unix domain socket connections only
local all all trust
# IPv4 local connections:
host all all 127.0.0.1/32 trust
# IPv6 local connections:
host all all ::1/128 trust
by replacing the text in the last column to “trust” as it is here (that’s the insecure part!). Then restart the postgresql server:
/etc/init.d/postgresql restart
Then, switch users to the “postgres” user and create a new user called “gmod”:
su - postgres
createuser gmod
Shall the new role be a superuser? (y/n) y
exit # to leave postgres user shell
exit # to leave root shell
Install DBIx::DBStag
This is a perl module that can only be installed after PostgreSQL is configured, so it is installed now. First, create a database called “test”:
createdb test
Then install via the cpan shell:
cpan
cpan> install DBIx::DBStag
Note that installing via the cpan shell is difficult if you typically use cpan as root, like “sudo cpan”. If instead you use cpan as a regular user but have it configured to do “sudo make install” and “sudo ./Build install” it is easy and works correctly.
GMOD Components
Chado Install and Load
Get Chado from SourceForge; point a browser at
http://sourceforge.net/projects/gmod/files/gmod/chado-1.11/chado-1.11.tar.gz/download
and extract the files:
cd ~/Downloads
tar zxvf chado-1.11.tar.gz
cd chado-1.11
Set up some environment variables:
vi ~/.bashrc
and add these lines to the bottom:
export GMOD_ROOT=/usr/local/gmod
export CHADO_DB_NAME=chado
export CHADO_DB_USERNAME=gmod
save .bashrc and source it so that the values are available in the shell:
source ~/.bashrc
Now to install Chado:
perl Makefile.PL
Accept all of the defaults except for the “default organism” question. Put “pythium” here.
make
sudo make install
make load_schema #ignore the error about a chado database not existing
make prepdb
make ontologies
answer with 1,2,4
Add our organism to the database:
psql chado
psql> INSERT INTO organism ( abbreviation, genus, species, common_name)
VALUES ('P.ultimum','Pythium','ultimum','pythium');
Make a database dump that saves progress to this point:
pg_dump chado | bzip2 -c > ontologies_only_dbdump.bz2
Loading Data
Note that if MAKER is installed locally, it provides a tool to automatically load data from it straight into Chado. Since we don’t have that, we’ll load “by hand”.
Go to where the data is:
cd ~/Downloads/3263.maker.output/
There are several files here, but the one we are interested in at the moment is 3263.all.gff, which has all of the “raw material” annotations made by MAKER as well as the final gene calls. We’ll load these into Chado using a tool that came with Chado:
gmod_bulk_load_gff3.pl -a --noexon -g 3263.all.gff
where the -a tells the loader that these are computational results (as opposed to human-curated annotations), and the –noexon tells the loader not to create exon features that correspond to the CDS features, because the exon features are already present in the GFF. This load takes a few minutes.
GBrowse Installation
GBrowse can be installed directly from the cpan shell like several of the GBrowse prerequisites were installed:
cpan
cpan> install Bio::Graphics::Browser2
Accept all of the defaults when asked questions.
Get a config file
Install Samtools
cd ~/Downloads
wget -O samtools.0.16.tar.bz2 http://sourceforge.net/projects/samtools/files/samtools/0.1.16/samtools-0.1.16.tar.bz2/download
tar jxvf samtools.0.16.tar.bz2
cd samtools-0.1.16
sudo apt-get install ncurses-dev
make #after adding fPIC to Makefile
Install Bio::DB::Sam
cpan
cpan> install Bio::DB::Sam
answer "/home/gmod/Downloads/samtools-0.1.16" for the location of bam.h
BAM and FASTA files placed in /var/www/gbrowse2/databases/pythium/
With all of the pieces in place, now we can go to http://localhost/cgi-bin/gb2/gbrowse/pythium (or http://gmodags.bx.psu.edu/cgi-bin/gb2/gbrowse/pythium during the workshop).
JBrowse Installation
Get JBrowse and unpack:
wget http://jbrowse.org/releases/jbrowse-1.2.1.zip
unzip jbrowse-1.2.1.zip
sudo cp -r jbrowse-1.2.1/ /var/www/jbrowse/
Get a conf file:
cd ~/Downloads/
wget https://gist.github.com/raw/1014946/65ab0c150984d7bed47150d82da5026d960406f3/pythium.conf
Setting up data:
cd /var/www/
sudo chown -R gmod:gmod jbrowse
cd jbrowse
bin/prepare-refseqs.pl --conf ~/Downloads/pythium.conf --refs scf1117875582023
This gets the “reference sequence”, that is, the contig we’re working on. At this point, visiting the jbrowse url (http://gmodags.bx.psu.edu/jbrowse) would show an empty jbrowse user interface, though if you zoom all the way in, you’d see DNA residues.
Now, get more data:
bin/biodb-to-json.pl --conf ~/Downloads/pythium.conf
This talks to the Chado database and extracts all of the evidence and predictions that were used and created in the MAKER analysis.
Now that JBrowse has extracted all of the data, we can go to http://localhost/jbrowse (or http://gmodags.bx.psu.edu/jbrowse during the workshop).
Installing Galaxy
The default python on Ubuntu 11.04 is 2.7. We need 2.6 to run Galaxy. Using the instructions from the GetGalaxy wiki page, Python 2.6 was downloaded and added at the front of the path.
mkdir ~/galaxy-python
ln -s /path/to/python2.5 ~/galaxy-python/python
~/.bashrc was edited and these lines were added to the end.
# Use Python 2.6 for Galaxy
export PATH=~/galaxy-python:$PATH
Galaxy was then downloaded:
cd ~/Documents
mkdir work
cd work
hg clone http://bitbucket.org/galaxy/galaxy-dist
And we then customized the landing image for this conference. (Details are not important.)
And now we can start it:
cd galaxy-dist
sh run.sh
And Galaxy is now installed and running. Goto http://localhost:8080.
If time permits …
Community!
- Wiki
- Mailing Lists
- Meetings and other Events - Next meeting is October 2011 GMOD Meeting
- Training and Outreach
- …
Remember …
The one true url: