GMOD
MotifFinder.pm
MotifFinder.pm is a GBrowse plugin written by Xiaoqi Shi. It finds sequence specific motifs using Position Weight Matrix and display results graphically as tracks in the genome browser.
Please feel free to contact the author for help or more information. Follow this link for background reading of Position Weight Matrix
Contents
download the plugin
The plugin is installed on the GBrowse of WormBase&modENCODE, you may access it directory here: http://www.wormbase.org/db/gb2/gbrowse or http://modencode.oicr.on.ca/
If you want to install it on your own GBrowse, please contact the author for source code and then follow the instruction below:
- save both ‘motiffinder’ and ‘MotifFinder.pm’ under GBrwose plugin diretory(set the permission as executable).
- save ‘matrices.txt’(example of the PFM tables) under GBrowse conf directory
- include “MotifFinder” in your main GBrowse.conf.
- specify the matrix file name in your species *.conf
[MotifFinder:plugin]
matrix = matrices.txt
Then you should be able to run the plugin!
How to use MotifFinder plugin
Access The Plugin
- From GBrowse main page, the PrimerDesigner plugin, as well as other installed plugins, can be accessed via the upper right menu.
- In GBrowse, navigate to the genomic region you interested in, then select ‘Annotate Sequence Motif’ from the menu and click ‘Configure’
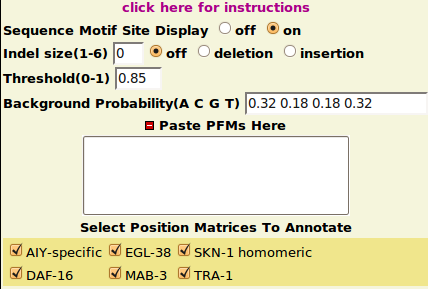
MotifFinder Parameters
- Reasonable default options are provided for each parameter.
- Threshold: a cutoff score between 0.8 to 1 is recommended.
- Background Probability: should be inputed in (A C G T) order.
- Indel Size: currently only small Indels(length under 6) can be handled.
Position Frequency Matrices
Existing PFMs were loaded from file ‘matrices.txt’ under GBrowse configuration directory, they are mostly curated PFMs from existing publications.
Click here for a list of all the available PFMs from WormBase
However, you can also add your own PFMs to the toggle section “Paste PFMs Here” in fasta format(arrange rows in A C G T order). e.g.
>name of the matrix ( the '>' sign is required !)
0 1 1 1 1 23 0 0 1 7 0 0 19
10 18 1 13 14 2 20 0 17 0 7 16 0
2 4 24 1 0 0 0 26 8 2 0 10 7
14 3 0 11 11 1 6 0 0 17 19 0 0
Indel Detection
User can search for sequence motifs that contain Indels up to certain length. This part hasn’t been fully tested and depends on future improvement.
Graphical Presentation
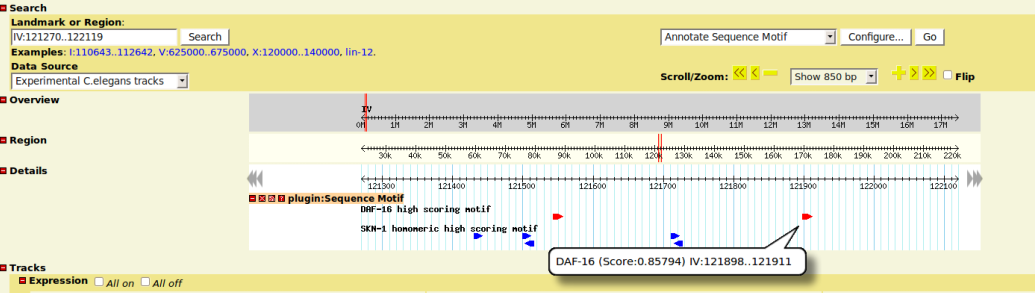
- each matching motifs is displayed as a glyph box on the tracks
- box arrow indicates the strand info
- move mouse on glyph will show you the computed similarity score and start/stop position of the motif
How is the motif predicted?
The problem is to find occurrences of known patterns(represented by position matrix) in new sequences.
Caculate Weight Score
Scoring function is the same as the TFBS Perl modules developed by Bergen University.
w = log2 ( ( f + sqrt(N) * p ) / ( N + sqrt(N) ) / 0.25 )
If we have PFM from TRANSFAC 7.0:
A 1 12 0 0 0 0 0 7 1 1 0 0 0 2 1
C 8 0 0 0 0 0 13 1 7 0 0 3 8 7 8
G 2 1 12 0 0 0 0 1 2 0 0 0 0 2 3
T 2 0 0 13 13 13 0 4 3 12 13 10 5 2 1
w - is a weight for the current nucleotide we are calculating
f - is a number of occurrences of the current nucleotide in the current column (e.g., “1” for A in column 1, “8” for C etc)
N - total number of observations, the sum of all nucleotides occurrences in a column (13 in this example)
p - [prior] [background] frequency of the current nucleotide; this one usually defaults to 0.25 (i.e. one nucleotide out of four)
Algorithms
- Backtrack: use recursive function to build all possible motifs, terminate recursion when an intermediate score is not reached.
- Brute-Force: calculate the similarity score across the whole region using a sliding window of motif size
This program uses a combined strategy by choosing between above two methods(depending length of the motif and cutoff score) to achieve faster computational speed .