GMOD
InterMine Presentation
This Wiki page is an edited version of Gos’s presentation
Contents
- 1 Background
- 2 Technical Overview
- 3 Loading Data
- 4 Code for Problem 2: Print gene annotation report
- 5 Quicksearch - Problem 4: find genes starting with x
- 6 Larger Query
- 7 Implications of Query Optimisation
- 8 Acknowlegements
Background
InterMine was developed as the generic underpinnings of the FlyMine Project
- Team of 7 FTE
- 5 developers, one sys admin,
- 1 biologist/ bioinformatician
- Java/ postgreSQL
- SVN repository: 125,000 lines of code + 57,000 lines of tests
- Under development since 2002
- In use by others in Cambridge, Edinburgh, Vienna… + modENCODE DCC if funded
- modENCODE/ Chado
Technical Overview
- Data model –> Java classes, relational schema, mappings through automatic code generation
- Custom Java object/relational system
- When we started, couldn’t select from multiple classes at one time using hibernate.
- Optimised for read-only performance
- Designed for big, complex queries, bulk data
- Performance optimisation
- Transparent query re-writing
- Web application - Struts/JSP/Ajax
Loading Data
- Read-only in production environment (therefore Problems 3 and 5 skipped)
- Load data from InterMine XML
- Parsers from standard formats
- e.g. UniProt, GFF3, PSI, FASTA
- Powerful integration system: coarse/fine grained data source priorities give load-order independence
Test problems
- Used SOFA as core data model - similar to Chado.
- Added Gene.description (absent from model), compiled, loaded data (here XML + FASTA), released webapp.
Example InterMine XML for Problem 1: load genes + annotation
<items>
<item id="0_3" class=”” implements="http://www.flymine.org/model/genomic#Gene">
<attribute name="identifier" value="xfile" />
<attribute name="description" value="A test gene for GMOD meeting" />
<reference name="organism" ref_id="0_1" />
<collection name="transcripts">
<reference ref_id="0_9" />
</collection>
</item>
<item id="0_1" class="" implements="http://www.flymine.org/model/genomic#Organism">
<attribute name="taxonId" value="7227" />
</item>
...
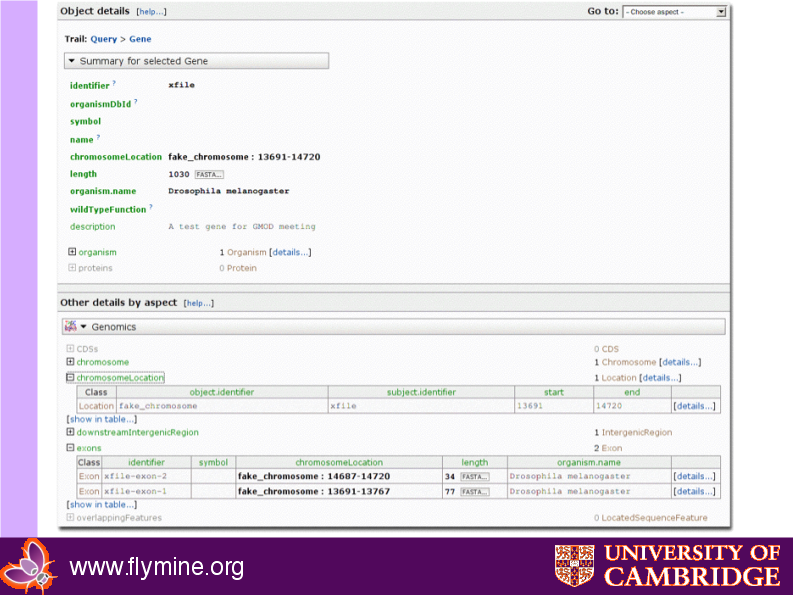
Resulting webapp object page
Code for Problem 2: Print gene annotation report
public class BakeOff {
public static void main(String[] args) throws Exception {
// code to get the "xfile" gene
ObjectStore os = ObjectStoreFactory.getObjectStore("os.production");
Query q = new Query();
QueryClass qcObj = new QueryClass(Gene.class);
q.addFrom(qcObj);
QueryField qf = new QueryField(qcObj, "identifier");
q.addToSelect(qf);
SimpleConstraint sc = new SimpleConstraint(qf, ConstraintOp.EQUALS, new QueryValue("xfile"));
q.setConstraint(sc);
System.err.println("query: " + q);
Results res = os.execute(q);
// a Results object is a List of Lists
List rr = (List) res.get(0);
Gene gene = (Gene) rr.get(0);
System.err.println ("symbol: " + gene.getIdentifier());
// a BioEntity in FlyMine has a collection of Synonym objects -
// we need Synonym.value for each Synonym
System.err.print ("synonyms: ");
Iterator synIter = gene.getSynonyms().iterator();
while (synIter.hasNext()) {
Synonym syn = (Synonym) synIter.next();
System.err.print (syn.getValue() + ' ');
}
System.err.println ("description: " + gene.getDescription());
// get the class name, but we already know that the gene is a Gene
System.err.println ("type: " + gene.getClass().getName());
// make a List from a the Set of exons for this Gene
List exons = new ArrayList(gene.getExons());
Exon exon1 = (Exon) exons.get(0);
Exon exon2 = (Exon) exons.get(1);
// get the start and end via the Location object
System.err.println ("exon1 start: " + exon1.getChromosomeLocation().getStart());
System.err.println ("exon1 end: " + exon1.getChromosomeLocation().getEnd());
System.err.println ("exon2 start: " + exon2.getChromosomeLocation().getStart());
System.err.println ("exon2 end: " + exon2.getChromosomeLocation().getEnd());
// write out the first cds
List cdss = new ArrayList(gene.getCDSs());
FlyMineSequence flymineSequence = FlyMineSequenceFactory.make((CDS) cdss.get(0));
// use BioJava to output the sequence
Annotation annotation = flymineSequence.getAnnotation();
annotation.setProperty(FastaFormat.PROPERTY_DESCRIPTIONLINE,
gene.getIdentifier() + " cds");
SeqIOTools.writeFasta(System.err, flymineSequence);
}
}
Quicksearch - Problem 4: find genes starting with x
Java API
Query q = new Query();
QueryClass qcObj = new QueryClass(Gene.class);
q.addFrom(qcObj);
q.addToSelect(qcObj);
QueryField qf = new QueryField(qcObj, "identifier");
SimpleConstraint sc = new SimpleConstraint(qf, ConstraintOp.MATCHES, new QueryValue("x-%"));
q.setConstraint(sc);
IQL
SELECT DISTINCT a1_.identifier AS a2_ FROM org.flymine.model.genomic.Gene AS a1_ WHERE a1_.identifier LIKE 'x-%'
Perl API
my $genes = InterMine::Gene::Manager->get_genes(query => [
identifier => { like => 'x-%' },],);
Larger Query
Within FlyMine: For one or more genes report:
- Gene, Transcripts, Exons, Chromosomal Locations, Lengths
- Query joins 7 classes
- all are on select list of query
- many more tables than classes are joined
- Performance:
- One gene:
- 2 rows in ~2 seconds
- All genes, all organisms
- ~300,000 rows in 36 seconds (without using pre-computation to enhance performance)
- ~300,000 rows in ~1 second (using pre-computation)
- One gene:
Implications of Query Optimisation
- Performance optimisation not tied to schema design
- Can adapt performance optimisation to usage of live database
- Template queries pre-computed
- ~40 template queries run per gene details page - renders in seconds
Acknowlegements
- Richard Smith
- Kim Rutherford
- Matthew Wakeling
- Xavier Watkins
- Julie Sullivan
- Rachel Lyne
- Hilde Janssens
- François Guillier
- Philip North
- Tom Riley
- Peter Mclaren
- Mark Woodbridge
- Debashis Rana
- Wenyan Ji
- Markus Brosch
- Florian Reising
- Andrew Varley
- Gos Micklem
InterMine/FlyMine are funded by the Wellcome Trust (grant no. 067205), awarded to M. Ashburner, G. Micklem, S. Russell, K. Lilley and K. Mizuguchi.