GMOD
Gbol
This is a scratch page for thoughts on development of the GMOD Bio Object Layer (GBOL).
== Note: This project is no longer active. ==
Contents
- 1 Object Layer Design Goals
- 2 Diagrams
- 3 Simple Object Layer
- 4 Bio Object Layer
- 5 Configuration Adaptor Thoughts
- 6 Analyses
- 7 Annotation Properties
Object Layer Design Goals
- Provide a simple object layer for manipulating generic genomic features, their locations, annotation properties, and supporting analyses. This object layer will essentially mimic the structure of GMOD’s Chado schema.
- Provide a set of biologically contextual objects that are extensions of the abstract layer and that have type-specific convenience functions and properties.
- The simple / bio object layers should be able to be used independently of any connection to a data store.
- Provide a factory interface for the fetching / storing of GBOL objects, as well as implementations that work with the Chado Schema and ChadoXML files.
- EL: Make sure that the biological layer adheres to Chado best practices in how it represents the data. We need to start standardizing use.
Diagrams
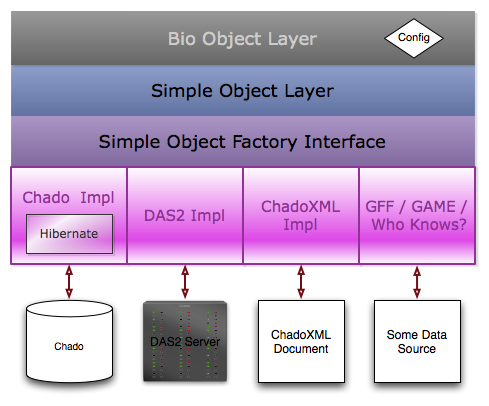
- The bio object layer contains classes that are extensions of the Simple Object layer classes. for instance, the Gene bio object class extends the simple object layer Feature class and has convenience methods like getTranscripts(). The feature set in the constructor by a configuration adaptor.
- The simple object layer is made of of classes that are essentially a transformation of the Chado Schema. For instance, there’s a CV class, CVTerm class, Feature class, etc.
- The simple object factory interface declares methods for fetching and saving simple objects to and from data stores. Initially, I’d like to implement that against chado & chado xml.
Simple Object Layer
As stated above, the simple object layer is essentially the chado schema in java object form.
- Random thoughts….
- In my first go around, I calculated equality and hascode based on the property values that are specified as the unique constraints of a table. For instance, equality of a Feature Object is based on organism, type, and uniquename, This might be unexpected behavior if people expect equality / hashcode to be evaluated based on all object fields instead of a subset.
- Definitely want to go for the schema-driven class generation approach if the tools are good enough.
Bio Object Layer
These BioObjects will be extensions of the simple object class Feature. We’ve discussed the idea of having some sort of “configuration adaptor” assigned to each bio object instance that would essentially specify the object’s type, detail how other bio objects are related to it, and tell what sorts of features to save should the object be written to a data store. See Configuration Adaptor Thoughts for more details.
Central Dogma Model thoughts:
- I think there’s a significant amount of confusion (or ambiguity) in the way the molbio central dogma is represented between MODs (and Apollo). Personally, I like the using a flybase-style protein feature who’s coordinates denote the start and stop of translation of a protein within a transcript, but the name protein is sort of a misnomer and a protein shouldn’t have a feature loc on a chromosome arm. So what about using a CDS feature to denote the boundaries of translation within a transcript? It could work just like the flybase protein feature - not multiple CDS regions on a per-exon basis but rather a single feature who’s coordinates dictate start and stop of translation within a transcript. It’s residues will be nucleotides and can be translated to get the polypeptide residues. There can be a polypeptide feature also, but it’s related to the CDS feature, not the transcript.
- Possible biological model: Gene->Transcript->(Single) CDS ->Polypeptide. The CDS feature would be a single feature that would denote the start and stop of translation within the transcript and would have coordinates relative to the genome or transcript. Genomic coordinates makes it easiest to calculate exon cds regions & urs. Transcript-relative coordinates seem most technically correct. The polypeptide would have no feature coordinates relative to the genome.
Extended Objects and methods:
- SO:0000704: Gene
- Thoughts:
- Methods:
- List<Transcript> getTranscripts(): Returns all transcripts related to a gene.
- boolean addTranscript(Transcript): Associates a transcript with a gene. Updates gene boundaries if necessary.
- List<Exon> getExons(): Returns all gene exons ordered by increasing start location.
- SO:0000673: Transcript
- Thoughts:
- Most of the objects involved in the CDS_region/ coding / UTR related functions could be calculated on the fly.
- Methods:
- Gene getGene(): returns gene that transcript is part of.
- List<Exon> getExons(): Returns transcript exons ordered by increasing start location.
- List<Exon> getCodingExons(): Returns transcript exons that contain coding sequence.
- List<Exon> getFivePrimeUTRExons(): Returns transcript exons containing 5’ UTR sequence.
- List<Exon> getThreePrimerimeUTRExons(): Returns transcript exons containing 3’ UTR sequence.
- boolean addExon(Exon): Associates an exon with a transcript. Updates transcript / gene boundaries if necessary.
- List<FivePrimeUTR> getFivePrimeUTRs(): Returns FivePrimeUTR features for a transcript. Should this actually be FivePrimeUTR_region?… see below.
- List<ThreePrimeUTR> getThreePrimeUTRs(): Returns ThreePrimeUTR features for a transcript. Ditto ThreePrimeUTR_region.
- List<Intron> getIntrons(): Returns intron features for a transcript.
- CDS getCDS() returns a single CDS feature for a transcript.
- List<CDS> getCDSRegions(): Returns CDS_region features for a transcript.
- Protein getProtein(): Not sure about this one. It’d be nice to assume that there was a single protein for any protein-coding transcript. How many people have multiple proteins associated with a single transcript?
- Thoughts:
- SO:0000147: Exon
- Thoughts:
- Methods:
- gene getGene(): returns gene that exon is part of.
- List<Transcript> getTranscripts: returns transcripts that exon is part of.
- boolean containsCDS(): returns true if exon contains coding sequence, false otherwise.
- boolean containsUTR(): returns true if exon has any UTR, false otherwise.
- boolean contains5primeUTR(): returns true if exon contains 5primeUTR, false otherwise.
- boolean contains3primeUTR(): returns true if exon contains 3primeUTR, false otherwise.
- CDSRegion getCDSRegion(): Returns CDSRegion compoenent of an exon if it exists.
- 5primeUTR get5primeUTR(): Returns 5primeUTR component of an exon if it exists.
- 3primeUTR get3primeUTR(): Returns 3primeUTR component of an exon if it exists.
- SO:0000188: Intron
- Thoughts:
- Methods:
- transcript getTranscript(): returns transcript that intron is a part of.
- SO:0000204: FivePrimeUTR:
- Thoughts:
- The SO definition indicates that it’s a contiguous sequence, but we want separate 5’ UTR pieces. So maybe a new FivePrimeUTR_region SO term is called for?
- Methods:
- transcript getTranscript(): returns transcript that UTR is a part of.
- exon getExon(): returns
- Thoughts:
- SO:0000205: ThreePrimeUTR:
- Thoughts:
- Same as FivePrimeUTR- Need a ThreePrimeUTR_region in so?
- Methods:
- transcript getTranscript(): returns transcript that UTR is a part of.
- Thoughts:
- SO:0000316: CDS
- Thoughts:
- The idea of having a cds feature define the translational boundaries of a transcript actually makes more sense than using a protein feature. So maybe the model should look like gene->transcript->cds->protein. The CDS feature would have nucleotides as residues and featurelocs that define the translational boundaries of a related protein feature.
- Methods:
- transcript getTranscript(): returns transcript that CDS is a subsequence of.
- String translate(): returns the translated peptide sequence of the CDS.
- Thoughts:
- SO:0000851: CDSRegion
- Thoughts:
- First, I don’t really see the utility of CDS_region features - they seem like a waste of space and all the information that they contain can simply be derived from single flybase-style protein feature (with translational start / stop coordinates as its featureloc). Actually… that should be a CDS feature instead of a protein. Despite this, it appears as though a number of MODs and Ensembl too maybe rely on CDS_region-style features (often just called ‘CDS’ features) so GBOL should support them to some extent.
- Methods
- transcript getTranscript(): returns transcript that CDSRegion is a part of.
- exon getExon(): returns the exon that the CDSRegion is a part of.
- Thoughts:
- SO:0000104: Polypeptide
- Thoughts:
- Should this even have a featureloc if the CDS feature is used to define translational boundaries?
- Should this contain the translated stop codon (*) from the CDS sequence?
- Methods:
- transcript getTranscript(): returns the transcript the the polypeptide derives from.
- CDS getCDS(): returns the CDS feature that the polypeptide is translated from.
- Thoughts:
Others….
- SO:0005836: RegulatoryRegion
- SO:0000188: Intron
- SO:0000105: ChromosomeArm
- SO:0000148: Supercontig
Configuration Adaptor Thoughts
Hmmm….
Analyses
I’d like a bioperl set of objects that allow you to traverse sequence alignments, hits, & HSPs. Certainly, people are always asking how to get analyses in and out of Chado. I think this will involve an extension of the Feature class like the bio object layer. Err… It probably should be part of the bio object layer.
Annotation Properties
Is there a controlled vocabulary for annotation properties? We need to specify conventions for things like, storing feature comments and comment properties. How about annotation evidence structure and confidence codes from GO?