GMOD
GBrowse syn Database
GBrowse_syn is a GBrowse based synteny viewer. This page describes the database that GBrowse_syn uses, and how to get syntenic data into that database.
- Sample data and configuration files can be downloaded from the GMOD FTP site; the sample data are for two rice species (courtesy of Bonnie Hurwitz)
Contents
Example Alignment Data
The sample below is in CLUSTALW format. Other formats are also supported (see below).
NOTE: The sequence naming convention “species-seqid(strand)/start-end” shown in the example below contains meta-data about the alignment that is essential for the data to be loaded correctly with strand and coordinate information
CLUSTAL W(1.81) multiple sequence alignment
c_briggsae-chrII(+)/43862-46313 ATGAGCTTCCACAAAAGCATGAGCTTTCTCAGCTTCTGCCACATCAGCATTCAAATGATC
c_remanei-Crem_Contig172(-)/123228-124941 ATGAGCCTCTACAACCGCATGATTCTTTTCAGCCTCTGCCACGTCCGCATTCAAATGCTC
c_brenneri-Cbre_Contig60(+)/627772-630087 ATGAGCCTCCACAACAGCATGATTTTTCTCGGCTTCCGCCACATCCGCATTCAAATGATC
c_elegans-II(+)/9706834-9708803 ATGAGCCTCTACTACAGCATGATTCTTCTCAGCTTCTGCAACGTCAGCATTCAGATGATC
****** ** ** * ****** ** ** ** ** ** ** ** ******* *** **
c_briggsae-chrII(+)/43862-46313 CGCACAAATATGATGCACAAATCCACAACCTAAAGCATCTCCGATAACGTTGACCGAAGT
c_remanei-Crem_Contig172(-)/123228-124941 AGCACAAATGTAATGAACGAATCCGCATCCCAACGCATCGCCAATCACATTCACAGATGT
c_brenneri-Cbre_Contig60(+)/627772-630087 CGCACAAATGTAGTGGACAAATCCGCATCCCAAAGCGTCTCCGATAACATTTACCGAAGT
c_elegans-II(+)/9706834-9708803 TGCACAAATGTGATGAACGAATCCACATCCCAATGCATCACCGATCACATTGACAGATGT
******** * ** ** ***** ** ** ** ** ** ** ** ** ** ** ** **
c_briggsae-chrII(+)/43862-46313 CCGGAGTCGATCCCTGAAT-----------------------------------------
c_remanei-Crem_Contig172(-)/123228-124941 ACGAAGTCGGTCCCTATAAGGTATGATTTTATATGA----TGTACCATAAGGAAATAGTC
c_brenneri-Cbre_Contig60(+)/627772-630087 ACGAAGTCGATCCCTGAAA---------TCAGATGAGCGGTTGACCA---GAGAACAACC
c_elegans-II(+)/9706834-9708803 ACGAAGTCGGTCCCTGAAC--AATTATTT----TGA----TATA---GAAAGAAACGGTA
** ***** ***** *
Note on formats
These example data are in clustalw format. The scripts used to process these data will recognize clustalw and other commonly used formats recognized by BioPerl’s AlignIO parser. This does not mean that clustalw is the program used to generate the alignment data.
- This particular alignment file in clustalw format was generated using a part of the compara pipeline.
- See this generalized hierarchical whole genome alignment workflow for general information on how whole genome alignment data can be generated.
{kind=link}
Note on the sequence ID syntax
The sequence ID is this clustal file is overloaded to contain information about the species, strand and coordinates. This information is essential:
rice-3(+)/16598648-16600199
The general format is species-refseq(strand)/start-end
species
name of species, genome, strain, etc (string with no ‘-‘ characters)
sequence
name of reference sequence (string with no ‘/’ characters)
(strand)
orientation of the alignment (relative to the reference sequence; + or
-)
start
start coordinate of the alignment relative to the reference sequence
(integer)
end
end coordinate of the alignment relative to the reference sequence
(integer)
Examples:
c_elegans-I(+)/1..2300
myco_bovis-chr1(-)/15000..25000
Loading the alignment database
Create a MySQL database
Before you load the database, make sure that a database of that name already exists; if not, create one from scratch using the following MySQL command:
mysql -uroot -ppassword -e 'create database my_database'
Loading from multiple sequence alignments
Multiple sequence alignments can be loaded directly into the GBrowse_syn alignment database with the script load_alignments_msa.pl. This script will process alignment data in a number of formats recognized by BioPerl, such as CLUSTAL and FASTA. Note that these are file formatting conventions used by a variety of different applications and use of one of the formats that do not imply that a particular program is the method of choice for generating your alignments. Whole genome alignments for multiple species are generally more complex than simple multiple sequence alignments with clustalw.
- More information on load_alignments_msa.pl
- See the GBrowse_syn page for more on whole genome alignment approaches
Loading from other sources
The script load_alignment_database.pl can be used to load the alignment database from a tab-delimited alignment data files (format described below). This format can either be an intermediate for parsed alignment data or can be used for data that does not come from multiple sequence alignments, for example gene orthology data, defined regions of co-linearity, etc. The tab-delimited format requires start and end coordinates for each reference sequence. Any features that have start end and strand information can be used.
- More information on load_alignment_database.pl
Data loading format
A tab-delimited intermediate format that encodes the alignment coordinates plus optional 1:1 mapping of coordinates within the alignment (to facilitate accurate grid-lines in GBrowse_syn). Each record is a single line (wrapped here for display only). Note that a reciprocal alignment is also created during database loading.
#species1 seqid1 start1 end1 strand1 reserved species2 seqid2 start2 end2 strand2 reserved \
# pos1-1 pos1-2 ... posn-1 posn-2 | pos1-2 pos1-1 ... posn-2 posn-1
c_briggsae chrI 1583997 1590364 + . c_remanei Crem_Contig24 631879 634679 - . \
1584000 634676 1584100 634584 (truncated...) | 631900 1590333 632000 1590233 (truncated ...)
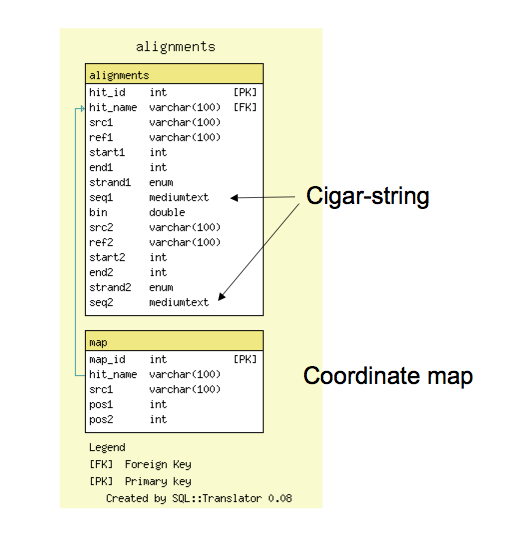
GBrowse_syn Database Schema
- The alignment database schema is very simple; it has a tables for all reciprocal ‘hits,’ or alignment features, and a table for (optional) 1:1 coordinate maps
- The alignments table contains coordinate information and also support cigar-line representations and the alignment to facilitate future reconstruction of the alignment within GBrowse_syn.