GMOD
GBrowse img
gbrowse_img - CGI script to generate genome images via the Generic Genome Browser
Contents
- 1 Description
- 2 Examples
- 3 CGI arguments
- 4 Generating from inside Gbrowse
- 5 Known Bugs
- 6 Author
- 7 Mediawiki Extension
- 8 See Also
Description
This CGI script is an interface to the Generic Genome Browser for the purpose of retrieving dynamic images of a region of the genome. It can be used as the destination of an <img> tag like this:
<img src="http://heptamer.tamu.edu/cgi-bin/gb2/gbrowse_img/MG1655/?name=NC_000913:3923000..3951000">
The script can also be used to superimpose one or more external features onto the display, for example for the purpose of displaying BLAST hits, an STS or a knockout in the context of the genome. It is designed to use a URL-based API to draw or embed GBrowse images without loading the full genome browser interface. gbrowse_img can be used to embed Gbrowse images in other webpages or to create high-resolution images appropriate for publications.
Examples
Gbrowse 2.26 with the Escherichia coli K-12 MG1655 genome (from bp 3923000 to 3950999) has been used to generate the following figures. Also note that the URLs have been encoded. Please be aware that your version of Gbrowse may operate slightly different based on its version.
Simple Example
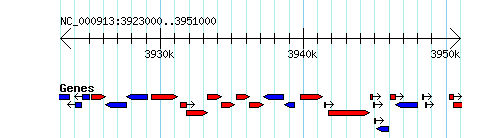
More complex
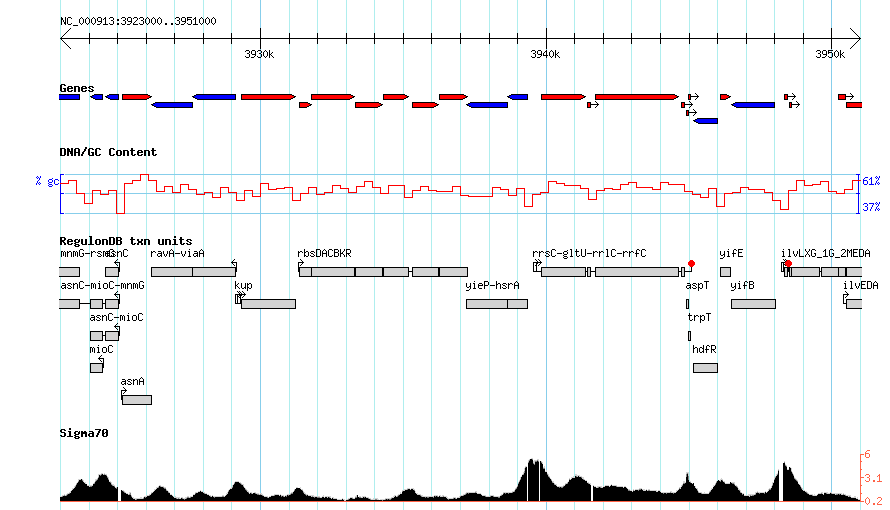
Listing Sources
You can get a list of sources (genomes, chromosomes, etc.), by setting the list parameter to sources like so:
http://heptamer.tamu.edu/cgi-bin/gb2/gbrowse_img/MG1655/?list=sources
## Sources
ATCC_8739
BL21
BL21_DE3
BL21_Gold_DE3
BW2952
DH10B
MG1655
etc.
Listing Types
To get a list of available tracks (the type parameter), set the list parameter to types like so:
http://heptamer.tamu.edu/cgi-bin/gb2/gbrowse_img/MG1655/?list=types
## Feature types for source MG1655
Genes Genes default
TranslationF 3-frame translation (forward)
TranslationR 3-frame translation (reverse)
DNA DNA/GC Content
Protein
rRNA_Operons
RegulonDBtu RegulonDB txn units default
Cryptic_Prophage cryptic prophage default
etc.
CGI arguments
The script recognizes the following CGI arguments, which can be passed either as GET or POST argument=value pairs. Argument pairs must be separated by semicolons (preferred) or by ampersands. Many of the options have one-letter aliases that can be used to reduce URL lengths.
| Argument | Alias | Description |
|---|---|---|
| name | q | genomic landmark or range |
| dbid | database ID for disambiguating names | |
| type | t | tracks to include in image |
| width | w | desired width of image |
| options | o | list of track options (compact, labeled, etc) |
| abs | b | display position in absolute coordinates |
| add | a | added feature(s) to superimpose on the image |
| style | s | stylesheet for additional features |
| keystyle | k | where to place the image key |
| overview | force an overview-style display | |
| flip | f | flip image left to right |
| grid | turn grid on (1) or off (0) | |
| embed | generate full HTML for image and imagemap for use in an embedded frame | |
| format | format for the image (use “SVG” for scaleable vector graphics) | |
| list | get certain types of configuration information | |
| source | database name |
The arguments are explained in more detail here:
name / q
This argument specifies the region of the genome to be displayed.
Several forms are recognized:
- name=Landmark Display the landmark named “Landmark”. Valid landmark names include chromosomes, contigs, clones, STSs, predicted genes, and any other landmark that the administrator has designated. Be careful when fetching large landmarks such as whole chromosomes!
- name=Landmark:start..end Display the region between start and end relative to “Landmark”.
- name=Class:Landmark Display “Landmark”, restricting to a particular class, such as “PCR_Product”. The list of classes is under the control of the database administrator and is not yet available through this interface.
- name=Class:Landmark:start..end As above, but restricted to the designated range.
If you use multiple name options, then this script will generate an overview image showing the position of each landmark. The alias “q” can be used to shorten the length of the URL.
dbid
If the data source contains multiple defined databases, this argument is
required to uniquely identify landmarks that may appear in multiple
databases under different names. If not present, the standard search
algorithm is used. Use the symbolic database name indicated in the
source configuration file. For example if the database stanza is
“[scaffolds:database]” then pass “dbid=scaffolds”.
type /t
This argument lists the feature types to display. The value of this
argument is a list of track names separated by spaces (“+” characters
when L-escaped). For example:
http://www.wormbase.org/db/seq/gbrowse_img/elegans?name=mec-3&type=tRNA+NG+WABA+CG+ESTB
Multiple type= arguments will be combined to form a single space-delimited list. The alias “t” can be used to shorten the length of the URL.
If the track name has a space in it, put quotes around the name:
type="microbe tRNA"+NG+WABA+CG+ESTB
width / w
Width of the desired image, in pixels.
options / o
A space-delimited list (“+” characters when URL-escaped) of
mnemonic/option pairs describing how features should be formatted.
Options are integers from 0 to 3, where 0=auto, 1=compact, 2=expanded,
3=expanded and labeled. For example, to specify that the tRNA and NG
tracks should always be expanded and labeled, but that the WABA track
should be compact, use:
options=tRNA+3+NG+3+WABA+1
The alias “o” can be used to shorten the length of the URL.
add / a
Superimpose one or more additional features on top of the view. Features
are specified as space (“+”) delimited lists in the following format:
add=Landmark+Type+Name+start..end,start..end,start..end
“Landmark” is the landmark name, “Type” is a descriptive type that will be printed in the image caption, “Name” is a name for the feature to be printed above it, and start..end is a comma-delimited list of ranges for discontinuous feature. Names that contain white space must be quoted, for example “BLAST hit”. Note that this all has to be URL-escaped, so an additional feature named “Your sequence”, type “Blast Hit”, that is located on chromosome III in a gapped range between 20000 and 22000, will be formatted as:
add=III+%22Blast%20Hit%22+%22Your%20Sequence%22+20000..21000,21550..22000
One or both of the type and name can be omitted. If omitted, type will default to “Your Features” and the name will default to “Feature XX” where XX is an integer. This allows for a very simple feature line:
add=III+20000..21000,21550..22000
Multiple add= arguments are allowed. The alias “a” can be used to shorten the length of the URL.
style
The style argument can be used to control the rendering of additional
features added with “add”. It is a flattened version of the style
configuration sections described in
this document. For example, if
you have added a “Blast Hit” annotation, then you can tell the renderer
to use a red arrow for this glyph in this way:
style=%22Blast%20Hit%22+glyph=arrow+fgcolor=red
keystyle / k
Controls the positioning of the track key. One of “right”, “left”,
“between” (default) or “bottom”
overview
Ordinarily the image will show the detail panel if the query region
corresponds to a single region, and the overview panel if multiple
regions match (or if a region that is too large to show matches).
Setting overview=1 will force the overview to be shown in all cases.
flip / f
Flip the image left to right. Arguments are 0=don’t flip (default), and
1=flip.
embed
Generate image and a corresponding HTML imagemap in a form suitable for
embedding into a frame.
format
Specify the format for the image file. Either “GD” (the default) or
“GD::SVG” for scaleable vector graphics.
list
If this argument is present, it will cause the script to dump out
various types of information in plain text form. Currently the two
values for this argument are sources, to dump out the list of data
sources, and types, to dump out the list of connfigured types. For
list=sources, the script will return a simple text list of the data
source names. For list=types, the script will return a three-column
tab-delimited list giving the track names and feature types
corresponding to the currently-selected data source. The format is as
follows:
Mnemonic <tab> Full description of feature <tab> [default]
The third column contains the word “default” if the track will be shown by default when no type argument is provided.
source
This argument specifies the data source for the images. The list of
sources can be found using list=sources. See also the
GBrowse_2.0_HOWTO#Configured_Data_Source_Sections configured data
source
sections
in the GBrowse 2 guide.
h_feat
The name of a feature to highlight in the format
feature_name@color_name
Example:
h_feat=SKT5@blue
You may omit “@color”, in which case the highlight will default to yellow. You can specify multiple h_feat arguments in order to highlight several features with distinct colors.
h_region
The name of a region to highlight in a solid background color, in the
format sequence_name:start..end@color_name
Example:
h_region=Chr3:200000..250000@wheat
You may omit “@color”, in which case the highlighted region will default to lightgrey. You can specify multiple h_region arguments in order to highlight several regions with distinct colors.
Image-maps
If you wish to associate the image with an imagemap so that clicking on a feature takes the user to the destination configured in the gbrowse config file, you may do so by placing the URL in an <iframe> section and using the embed=1 flag:
<iframe src="http://localhost/cgi-bin/gbrowse_img/elegans?name=B0001;embed=1" width="100%" height="250">
<img src="http://localhost/cgi-bin/gbrowse_img/elegans?name=B0001"/>
</iframe>
Placing an <img> tag inside the <iframe> tag arranges for older browsers that don’t know about iframes to display the static image instead. You may need to adjust the width and height attributes in order to avoid browsers placing scrollbars around the frame.
Generating from inside Gbrowse
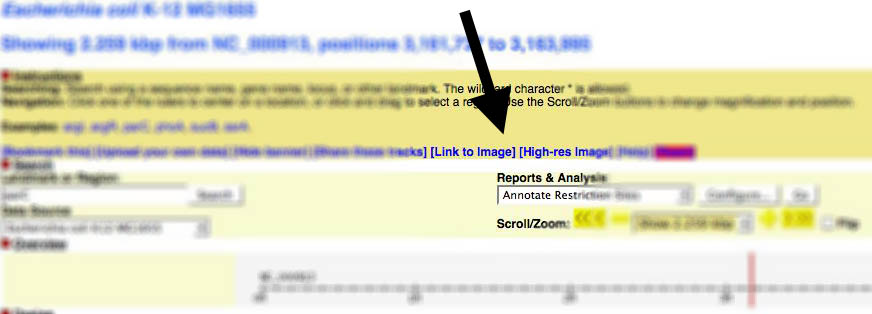
Gbrowse v1.x
You can find a link to generate images from within Gbrowse near the top of the page:
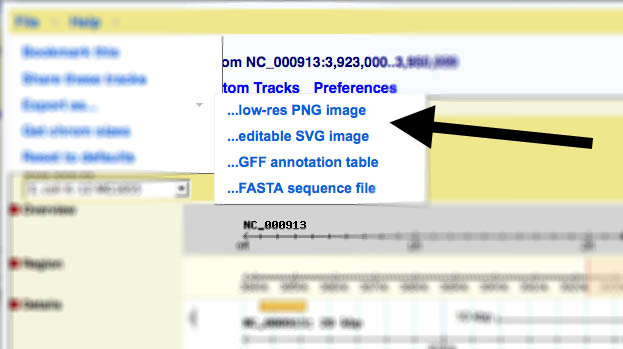
Gbrowse v2.x
In version 2, the link to generate images has been moved to the file menu:
Known Bugs
The cookie that stores the configuration options for plugins does not transfer from gbrowse to gbrowse_img, so tracks generated by annotation plugins, such as the Restriction site annotator, will not display correctly when the image URL is generated on one machine and then viewed on another. Uploaded files will transfer correctly, however.
Author
[mailto: lstein@cshl.org Lincoln Stein] Copyright (c) 2002-2004 Cold Spring Harbor Laboratory
This library is free software; you can redistribute it and/or modify it under the same terms as Perl itself.
Mediawiki Extension
The GbrowseImage extension for Mediawiki will display an image (rendered by gbrowse_img) in a wiki-page.