GMOD
Chado Sequence Module
Contents
- 1 Introduction
- 2
Features
- 2.1 Names of Features
- 2.2 Feature Synonyms
- 2.3 Feature Locations
- 2.4 Feature Rank
- 2.5 Extensible Feature Properties
- 2.6 Linking Features to External Databases
- 2.7 Feature Annotations
- 2.8 Relationships Between Features
- 2.9 Compliance
- 3
Tables
- 3.1 Table: feature
- 3.2 Table: feature_cvterm
- 3.3 Table: feature_cvterm_dbxref
- 3.4 Table: feature_cvterm_pub
- 3.5 Table: feature_cvtermprop
- 3.6 Table: feature_dbxref
- 3.7 Table: feature_pub
- 3.8 Table: feature_pubprop
- 3.9 Table: feature_relationship
- 3.10 Table: feature_relationship_pub
- 3.11 Table: feature_relationshipprop
- 3.12 Table: feature_relationshipprop_pub
- 3.13 Table: feature_synonym
- 3.14 Table: featureloc
- 3.15 Table: featureloc_pub
- 3.16 Table: featureprop
- 3.17 Table: featureprop_pub
- 3.18 Table: synonym
Introduction
A central module in Chado is the sequence module. The fundamental table within this module is the feature table, for describing biological sequence features. Chado defines a feature to be a region of a biological polymer (typically a DNA, RNA, or a polypeptide molecule) or an aggregate of regions on this polymer. As the term is used here, region can be the entire extent of the molecule, or a junction between two bases. Features can be typed according to an ontology, they can be localized relative to other features, and they can form part-whole and other relationships with other features.
You may find these related documents useful:
- Chado Manual
- Chado Best Practices - many issues specific to the Sequence module are discussed
- Chado FAQ
- Introduction to Chado
- Chado cv module - the Sequence module makes extensive use of controlled vocabularies
Features
Chado does not distinguish between a sequence and a sequence feature, on the theory that a feature is a piece of a sequence, and a piece of a sequence is a sequence. Both are represented as a row in the feature table.
There are many different types of features. Examples include gene, exon, transcript, regulatory region, chromosome, sequence variation, polypeptide, protein domain and cross-genome match regions. Chado does not have a different table for each kind of feature; all features are stored in the feature table.
Feature types are taken from the Sequence Ontology controlled vocabulary (see also Controlled Vocabulary module, also known as cv). Types of feature are differentiated using a type_id column, which is a foreign key to the cvterm table in the cv (ontology) module, described here. This allows us to type features according to the Sequence Ontology. The use of ontologies to type tables gives Chado a subtyping mechanism, which is absent from the standard relational model. For example, SO tells us that mRNA and snRNA are different kinds of transcript. This is discussed in more in the next section. For the purposes of discussion in this document, it can be assumed that any reference to genes, exons, polypeptides, SNPs, chromosomes, transcripts and various kinds of RNAs and so on refers to features of that Sequence Ontology type.
A selection of Chado-relevant types from SO are shown below:
| SO Term | SO id | | ——- | —– |
| Exon | SL:0000025 | | Intron | SL:0000027 | | mRNA | SL:0000037 | | miRNA | SL:0000044 | | regulatory_element | SL:0000052 | | transcription_factor_binding_site | SL:0000054 |
Sequence Ontology Examples
The Chado feature table has a text-valued column named residues for storing the sequence of the feature. The value of this column is string of IUPAC symbols corresponding to the sequence of biochemical residues encoded by the feature. This column is optional, because the sequence of the feature may not be known. Even if the sequence of a feature is known, it may not be desirable to store it in the feature table, as it may be possible to infer the sequence from the sequence of other features in the database. For example, exon sequences are generally not stored, as these can trivially be inferred from the sequence of the genomic feature on which the exon is located. In contrast, mRNA and other processed transcript sequences are stored as it is less trivial and more computationally expensive to dynamically splice together the mRNA sequence.
It is important to realize that the existence of a row in the feature table does not necessarily imply that the feature has been characterized as a result of genome annotation. It is possible to have features of SO type gene for genes that have only been characterized through genetic studies, and for which neither sequence nor sequence location is known. This is in contrast to other feature schemas (such as GFF) in which it is not possible to represent features without representing a location in sequence coordinates. This design decision is crucial for the use of Chado as a database for integrating information about the same entity from multiple perspectives.
Because the sequence is stored as a column in the feature table rather than as an independent table, sequences cannot exist in the absence of a row in the feature table; sequences are dependent upon features. This is in contrast with almost all other genomics schemas that allow independent treatment of sequences and features. This design decision follows for both philosophical and prag- matic reasons. The feature table also contains columns seqlen and md5checksum, for storing the length of the sequence and the 32-character checksum computed using the MD5 [RL Rivest. RFC 1321: The md5 message-digest algorithm. Technical report, Internet Activities Board, April 1992.] algorithm. The length and checksum can be stored even when the residues column is null valued. The checksum is useful for checking if two or more features share the same sequence, without comparing the entire sequence string.
The existence of these columns means that this table is no longer in third normal form (3NF), which is usually a desirable formal property of relational database. On balance, the utility of these columns outweighs the disadvantages of violating 3NF. In practical terms, it means that the values of the residues, seqlen and md5checksum columns are interdependent and cannot be updated independently of one another.
The feature table has a Boolean valued column, is_analysis, indicating whether this is an annotation or a computed feature from a computational analysis. Annotations are features that are generated or blessed by a human curator, or in some cases by an integrated genome pipeline (for example, MAKER or DIYA) capable of synthesizing gene models and other annotations from in silico analyses. They constitute the definitive version of a particular feature, in contrast to the features generated by gene prediction programs and sequence similarity searches such as BLAST.
The feature table has a dbxref_id column that refers to a global, stable public identifier for the feature. This column is optional, because not all classes of features have such identifiers for example, features resulting from gene predictions and BLAST HSP features may be less stable and thus lack public identifiers. It is recommended that most annotated features have dbxref_ids. The organism_id column refers to a row in the organism table (defined in the organism module). This column is mandatory if the feature derives from a single organism.
Names of Features
The name and uniquename columns allow features to be labelled. The name column is optional, but it is recommended that all annotated features (as opposed to those that arise from purely computational methods) have names. The name should be a simple, concise, human-friendly display label (such as a gene or gene product symbol, as defined by the nomenclature rules of governing the organism). User interface software (such as GBrowse and Apollo) can use the name column for labelling feature glyphs in user displays. Uniqueness of name within any particular organism or genome project is a desirable characteristic, but is not enforced in the schema, since there are occasions where name clashes are unavoidable. In contrast, the uniquename column is required, and guaranteed to be unique when taken in combination with organism_id and type_id this is enforced by a constraint in the relational schema. The unique name may be human-friendly (for example, it can be the same as the name); however, it is not guaranteed to be so, and in general should not be displayed to the end user. Its use is mainly as an alternate unique key on the table .
The unique name normally conforms to some naming rule these rules may vary across chado instances, but they should all guarantee the uniqueness of the uniquename, organism id, type id triple.
Feature Synonyms
In addition to having a name or symbol, it is common for features such as genes to have multiple synonyms or aliases. These synonyms may exist due to different publications referring to the same gene with different symbols, or because one gene was once believed to be two or more separate genes. A common curation operation on genes is splitting and merging, which results in the creation of synonyms.
This is modelled in Chado with a synonym table and a feature_synonym linking table; thus multiple features can potentially share the same, and a single feature can be have multiple synonyms. Use of a synonym in the literature is indicated with a pub_id foreign key referencing the pub table (see the publications module), indicating historical provenance for the use of a synonym.
Feature synonyms are found by joining to feature_synonym and synonym. For example, here is a query to find gene by name or synonym:
SELECT feature_id FROM feature
WHERE name = 'name of interest'
UNION SELECT feature_id
FROM feature_synonym fs, synonym s
WHERE fs.synonym_id = s.synonym_id
AND s.name = 'name of interest'
AND fs.is_current;
Feature Locations
Features can potentially be localized using a sequence coordinate system. A relative localization model is used, so all feature localizations must be relative to another feature. Some features such as those of type chromosome are not localized in sequence coordinates. Locations are stored in the featureloc table, also part of this sequence module. Other non-sequence oriented kinds of localization (such as physical localization from in situ experiments, or genetic localizations from linkage studies) are modelled outside the sequence module (for example, in the expression module or map module).
A feature can have zero or more featurelocs, although it will typically have either one (for localized features for which the location is known) or zero (for unlocalized features such as chromosomes, or for features for which the location is not yet known, such as a gene discovered using classical genetics techniques). Features with multiple featurelocs will be explained later.
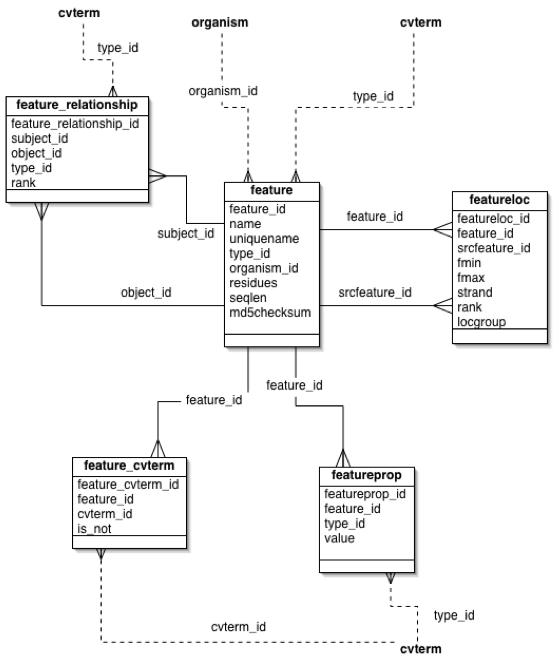
A featureloc is an interval in interbase sequence coordinates (see figure), bounded by the fmin and fmax columns, each representing the lower and upper linear position of the boundary between bases or base pairs, with directionality indicated by the strand column. Interbase coordinates were chosen over the more commonly used base-oriented coordinate system because they are more naturally amenable to the standard arithmetic operations that are typically performed upon sequence coordinates. This leads to cleaner and more efficient database coding logic that is arguably less prone to errors. Of course, interbase coordinates are typically transformed into the more common base-oriented system used by BLAST reports and so forth prior to presentation to the end-user.
The relational schema includes a constraint which ensures that fmin != fmax is always true, and any attempt to set the database in a state which violates this will flag an error .
As mentioned previously, a featureloc must be localized relative to another feature, indicated using the srcfeature_id foreign key column, referencing the feature table. There is nothing in the schema prohibiting localization chains; for example, locating an exon relative to a contig that is itself localized relative to a chromosome (see figure). The majority of Chado database instances will not require this flexibility; features are typically located relative to chromosomes or chromosomes arms. Nevertheless, the ability to store such localization networks or location graphs can be useful for unfinished genomes or parts of genomes such as heterochromatin, in which it is desirable to locate features relative to stable contigs or scaffolds, which are themselves localized in an unstable assembly to chromosomes or chromosome arms. Localization chains do not necessarily only span assemblies protein domains may be localized relative to polypeptide features, themselves localized to a transcript (or to the genome, as is more common). Chains may also span sequence alignments.
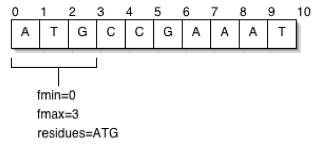
The Feature Location Graph
We will now present a short formal treatment of the properties of these hierarchies of localization using graph theory. This treatment can be ignored for the purposes of understanding the basics of the Chado schema; the end-user of the database will be entirely unaware of such technicalities. However, for the purposes of software engineering and ensuring interoperability between different Chado database instances and different applications, formal treatments such as these are an essential requirement for software specifications.
We can define a featureloc graph (LG) as being a set of vertices and edges, with each feature constituting a vertex, and each featureloc constituting an edge going from the parent feature_id vertex to the srcfeature_id vertex. The node is labeled with column values from the feature table, and the edge is labeled with column values from the featureloc table. The LG is not allowed to contain cycles, it is a directed acyclic graph (DAG). This includes self-cycles - no feature may be localized relative to itself.
The roots of the LG are the features that do not have featureloc rows, typically chromosomes or chromosome arms, although LG roots may also be unassembled contigs, scaffolds or features for which sequence localization is not yet known (such as genes discovered through classical genetics techniques). The leaves of the LG are any features that are not present as a srcfeature_id in any featurelocs row typically the bulk of features, such as genes, exons, matches and so on. The depth of a particular LG g, denoted D(g), is the maximum number of edges between any leaf- root pair. As has been previously noted, many Chados will have LGs with a uniform depth of 1. Such LGs are said to be simple and the features within them are said to be singletons. The maximum depth of all LGs in a particular database instance i is denoted LGDmax(i).
The schema does not constrain the maximum depth of the LG. This flexibility proves useful when applying Chado to the highly variable needs of multiple different genome projects; however, it can lead to efficiency problems when querying the database. It can also make it more difficult to write software to interoperate with the database, as the software must take into account different contingencies. We can solve this problem by collapsing the LG, in which a graph of arbitrary depth is flattened to a depth of 1, transforming or projecting featurelocs onto the root features (typically chromosomes or chromosome arms). The original featurelocs are left unaltered in the database, and additional redundant featurelocs between leaf and root features are added to the database. These new featurelocs are known as inferred featurelocs. In the schema inferred featurelocs are differentiated from direct featurelocs using the locgroup column. Direct (non-inferred) localizations are indicated by the locgroup column taking value 0, and transitive localizations are indicated by this column having value !0.
The terminology used above can be used to define specifications for applications intended to interoperate with the database. Certain kinds of features have paired locations. These include hits and high-scoring-pairs (HSPs) coming from sequence search programs such as BLAST, and syntenic chromosomal regions. These kinds of features have two featurelocs (in contrast to the usual 1) one on the query feature and one on the subject (hit) feature. We differentiate the two featurelocs with the rank column. A rank of 0 indicates a location relative to the query (as is the default for most features), and a rank of 1 indicates a location relative to the subject (hit) feature.
For multiple alignments (e.g. CLUSTALW results), this scheme is extended to unbounded ranks [0..n], with arbitrary ordering. Alignments are stored in the residue info column. CIGAR format is used for pairwise alignments.
Multiple featurelocs may also be required for features of type “sequence variant” (SO:0000109), indicating points or extents which vary between reference and non-reference sequences. From a modelling standpoint, variants are conceptually similar to alignments; with variants we are noting a difference as opposed to a similarity. Here a rank of zero indicates the wild-type (or reference) feature and a rank of one or more indicates the variant (or non-reference) feature, with the residue info column representing the sequence on wild-type and variant. A featureloc is uniquely identified by the feature_id, rank, locgroup triple. This means that no feature can have more than one featureloc with the same rank and locgroup. In other words, rank and locgroup uniquely identify a featureloc for any particular feature.
Feature Coordinates
Features are located relative to other features using the featureloc table rows. Features can be located on more than one sequence. For example, a BLAST hit HSP can be a feature of both the query and target sequences. To locate a feature, create a featureloc record with:
- srcfeature_id = the id of the sequence on which the feature is being located
- feature_id = the id of the feature being located
- strand is 1 for the positive strand, -1 for the negative, and 0 for both or indifferent.
- fmin, fmax – the minimum and maximum coordinates of the interval
- is_fmin_partial, is_fmax_partial = true if needed to indicate that the sequence is incomplete (e.g. for ESTs or EST assemblies which are known to not go all the way to the 3’ or 5’ end.)
- phase = 0, 1, or 2 – denotes phase of first base pair in a nucleotide feature with respect to a source protein, or the offset of the first nucleotide in its codon.
- rank, locgroup – these are used to organize groups of feature locations and can be ignored in simple cases (the details are discussed below).
Multiple Locations for a Feature
The ability to have multiple locations for a feature has many uses. For example one can locate a SNP, exon, or protein motif on the genome, on a transcript, and on a protein. A region of similarity between two sequences (HSP) can be located on both of them, so if either is viewed the “hit” is visible.
Difference Between the chado Location Model and Other Schemas
There is a crucial difference between the Chado location model and the sequence location model used in other schemas, such as GFF, GenBank, BioSQL, or BioPerl.
First, Chado is the only model to use the concept of rank and locgroup. Second, and perhaps more important, all these other models allow discontiguous locations (also known as “split locations”). These will be familiar to anyone who has inspected GenBank annotated DNA records for an organism that has introns within the transcripts; the transcript location is modelled as a sequence of non-contiguous intervals on the genome. The interval represents the location of an exon. For example:
/gene="Acph"
CDS join(914..1063, 1143..1241, 1297..1536, 1605..2054,
2667..2925, 3063..3172)
Although Chado allows a feature to have multiple locations, this is only with variable rank and locgroup and this is enforced by a uniqueness constraint in the relational schema. We made a conscious decision to avoid discontiguous locations, because the extra degree of freedom this affords results in either redundancies or ambiguities. Redundancies arise when exons are stored in addition to a discontiguous transcript, and ambiguities arise by virtue of the fact that explicit representation of the exons may be seen as optional. Ambiguities are undesirable as it makes it harder for databases to interoperate. The omission of discontiguous locations does not restrict the expressive capacity of Chado in any way, because any discontiguous location can be modelled as a collection of features with contiguous locations. For example, a transcript with a discontiguous location can be modelled as a collection of exons with contiguous featurelocs, and a transcript with a single contiguous featureloc representing the outer boundaries defined by the outermost exons.
Feature Rank
The rank field is used when a feature has more than 1 location, otherwise the default rank value of 0 is used. Some features have two locations, for example BLAST hits and HSPs: one location on the query, rank = 0, and one location on the subject, rank = 1.
Extensible Feature Properties
The feature table has a fairly limited set of columns for recording feature data. For example, there is no anticodon column for recording the RNA triplet for the adapter in a tRNA feature (all feature types, including tRNAs, are recorded as rows in the feature table). If we were to add columns such as anticodon then the number of columns in the table would become very large and difficult to manage; most would end up being nullable (for example, anticodon does not apply to non-tRNA features). This is because different organisms, different types of feature and different projects have differing needs regarding what extra data should be attached to any one feature. How then are we to attach both biologically relevant and project specific data to features?
Chado solves this by using an extensible mechanism for attaching attribute-value pairs to features via the featureprop table. The featureprop.type_id foreign key column references a property in the Sequence Ontology. The value text column stores the value filler for that property. Sets or lists of values for any property can be stored in the featureprop table, differentiated by the value of the rank column. Provenance for the featureprop assignment is stored using the featureprop_pub table in the publications module, allowing multiple publications to be associated with any one assignment.
Because featureprop values can be of an arbitrary size, they are modelled using a SQL TEXT type. This has some disadvantages from a query efficiency perspective.
Numeric values cannot be indexed correctly, and sorting the results of a query can only be done via a SQL casting operation, or in software outside of the database management system, either of which may result in poorer performance. This is one of several areas in Chado where performance has been traded in favour of a simpler, more abstract and generic model.
Linking Features to External Databases
Public database identifiers are stored in the dbxref table, which holds the database name, the accession number, and an optional version number. Note that this table holds accession numbers published internally by the Chado instance as well as by other databases. A feature can have a primary dbxref, which is linked directly from the feature table. It can also have additional secondary dbxref’s linked via feature_dbxref. A feature need not have a primary dbxref; e.g. computed features may be considered “lightweight” and not assigned accession numbers. Some groups may wish to set up a trigger to automatically assign primary dbxrefs to features of types that are locally accessioned; a sample trigger is provided with the schema.
Feature Annotations
Detailed annotations, such as associations to Gene Ontology (GO) terms or Cell Ontology terms, can be attached to features using the feature_cvterm linking table. This allows multiple ontology terms to be associated with each feature.
Provenance data can be attached with the feature_cvtermprop and feature_cvterm_dbxref higher-order linking tables. It is up to the curation policy of each individual Chado database instance to decide which kinds of features will be linked using feature_cvterm. Some may link terms to gene features, others to the distinct gene products (processed RNAs and polypeptides) that are linked to the gene features.
Annotations for existing features can also go into the featureprop
table using the Chado feature_property ontology
(defined in chado/load/etc/feature_property.obo) and the comment or
description terms as appropriate. The purpose of the feature property
ontology (and the related chado/load/etc/genbank_feature_property.obo
file) is to capture terms that are likely to appear in GFF
or GenBank sequence files. In theory there is no overlap between these
ontologies and the Sequence Ontology.
Relationships Between Features
Biological features are inter-related; exons are part of transcripts, transcripts are part of genes, and polypeptides are derived from messenger RNAs. Relationships between individual features are stored in the feature_relationship table, which connects two features via the subject_id and object_id columns (foreign keys referring to the feature table) and a type_id (a foreign key referring to a relationship type in an ontology, either SO, or the OBO relationship ontology, OBO-REL, indicating the nature of the relationship between subject and object features.
The core relationships between features are part-whole (part_of) or temporal (derives_from). Subject and Object describes the linguistic role the two features play in a sentence describing the feature relationship. In English, many sentences follow a subject, predicate, object syntax, and word order is important. To say that ”exons are part of transcripts” is the correct way to describe a typical biological relationship. To say ”transcripts are part of exons” is either grammatically or biologically incorrect.
We use this same terminology (which comes from RDF) again in the cv module. The collection of features and feature relationships can be considered as vertices and edges in a graph, known as the Feature Graph (FG). Example feature graphs are shown above and in the Introduction to Chado.
The FG is independent of the LG and in general the FG and the LG should have no edges in common. If there is a featureloc connecting two features, then the addition of a feature relationship between these same two features is redundant. The FG is required in order to query the database for such things as alternately spliced genes, exons shared between transcripts, etc.
Although the chado schema admits any FG, certain configurations are biologically meaningless, and should not be used. The FG can be constrained by the Sequence Ontology. Standardized FG structures are required for complex applications to be interoperable.
Unlike the LG, the FG may be cyclic, although cycles in the FG are not common. The subset of the FG corresponding to certain kinds of relationship may be acyclic for example, the subset of the FG connecting parts with wholes via part of must be acyclic.
Compliance
This section is not complete, it is in progress.
Chado uses a layered model - this is tried and tested in software engineering. Some generic software can be targeted at the lower layers and be guaranteed to work no matter what. Other more specific software needs a more tightly defined rigorous model and should be targeted at the upper layers.
We require validation software and more formal or computable descriptions of these layers and policies - for now natural language descriptions will have to suffice.
Chado Compliance Layers
Proposal for levels of compliance.
Level 0: Relational Schema
Level 0 conformance basically means the schema is adhered to. Obviously, this is enforced by the DBMS.
Layer 1: Ontologies
Level 1 conformance is minimal conformance to SO - all feature.types must be SO terms, and all feature relationship.types must be SO relationship types.
Level 2: Graph
Level 2 conformance is graph conformance to SO - all feature relationships between a feature of type X and Y must correspond to relationship of that type in SO; for example, mRNA can be part of gene, but mRNA can not be part of golden path region. [more detailed/formal explanation to come]. In practice Level 2 conformance may be undesirable, we may need to make modifications to SO.
Orthogonal to these layers are various additional policy decisions. Some of these are more tolerant of non-conformance than others. (there is also some overlaps with levels 1 and 2).
Examples: Current implementations
This section describes details of how different sites are using Chado. This is likely outdated information.
TIGR: Currently at level 0 conformance, though most (if not all) of the terms being used have an obvious counterpart in SO. Therefore these ”TIGR Ontology” terms are used in the answers to the SO-related questions that appear below. We plan on updating our terms with SO terms very soon.
SO terms used for Standard Central-dogma Gene Model
FlyBase: gene mRNA exon protein [other types are derivable].
TIGR: gene transcript CDS exon protein [though the strict answer is for any of these SO questions is ”none” since we do not yet meet level 1 conformance].
NOTE: we should be using ’polypeptide’ instead of ’protein’. For now, software should be tolerant of both these uses.
SO terms Used for Storing Alignments
FlyBase: match
TIGR: match
NOTE: we want to use the new more specific SO types for match set, match part, for hits and hsps respectively. For now, software should be tolerant of either usage.
TIGR: We’ve also extended the model for storing pairwise alignments to store multiple alignments. Each member of the alignment is featureloced to the ’match’ feature. We’ve used this representation to store paralogous/orthologous gene families.
feature_relationship Types
FlyBase: partof (for mRNA to gene and exon to mRNA) producedby (for protein to mRNA)
TIGR: part of (gene-assembly, exon-transcript, assembly-supercontig) produced by (protein- CDS, CDS-transcript, transcript-gene)
NOTE: this should be ”part of” and ”derived from” to conform to SO. Most read-only software should be able to safely ignore feature relationship.type anyway. Protein should be polypeptide - see note above
NOTE: the main difference between FB and TIGR here is that TIGR introduce an intermediate CDS feature between mRNA and protein.
featureloc Policy
FlyBase: all constituent parts of a central dogma gene model are located relative to the same srcfeature (the chromosome arm). No redundant locations (i.e. featureloc.group ¿ 0) are used.
TIGR: Redundant locations are used and indicated with featureloc.group ¿ 0.
NOTE: we want to allow some flexibility with this policy. We believe that the constituent parts linked located relative to the feature should always be followed. This can be stated more formally as:
IF X is linked to Y via feature_relationship
AND X is located relative to Z via featureloc.srcfeature_id
THEN Y must also be located relative to Z via featureloc.srcfeature_id
TIGR: We’ve followed this policy in adding a featureloc between the protein and genomic contig in our databases (such a featureloc does not appear in the Chado usage documents). This additional featureloc simplifies many queries, especially when looking at the genomic context of ’match’ features associated with proteins.
We should also expect that the fmin/fmax boundaries of a feature be defined the the outermost boundaries of the outermost constituent part features (this rule may require refinement when we have promoters, enhancers and so on - but for now we don’t).
As to what the srcfeature should be, it could be a contig, and assembly or a top-level locat- able feature such as chromosome or chromosome arm. Software should be tolerant of different choices here. Whilst it is generally always best to locate relative to the topmost feature (ie the arm/chromosome), sometimes this is not possible or desirable (eg low coverage, heterochromatin).
Non-central Dogma Gene Models
FlyBase: we store a lot of non-central dogma gene models; noncoding gene models and pseudogenes [need to fill in more details here].
TIGR: not many of these stored yet, save for a few pseudogenes and the occasional non-coding ORF.
Other Features
FlyBase: the FlyBase implementation includes many other feature types, including polyA site and se- quence variant [need to fill in details].
TIGR: using ’SNP’ in some databases.
Derivable Feature Types
FlyBase: derivable features (introns, UTRs, intergenic region) are not included. Feature typing is always done to the most specific, non-derivale level. For example, we never use types ”5 prime exon”, ”dicistronic gene”, ”coding exon” as these are always inferrable. We always use type ”gene” - the specific type of gene is inferred from the child type (mRNA, tRNA, snRNA, etc)..
TIGR: derivable features are not included. currently not storing any tRNAs or snRNAs.
NOTE: whilst it is perfectly permissable to include redundant derivable features (useful for warehouse-style querying), you should not write software that expects to find these if you want the software to work on different chado db instances.
Sequence Variants
FlyBase: these are included in chado, but they are lacking full detail.
TIGR: only SNPs so far. the SNPs currently being stored are computed from pairwise alignments of sequences already loaded into Chado, so each SNP feature is featureloc’ed to the appropriate place on each of the two sequences (rather than having one of the featurelocs ”dangling”, as indicated in some of the Chado usage documents.) featureloc.residue info is used to redundantly store the base referenced in each of the two sequences.
NOTE: variation features should specify the edit that makes one feature (such as the reference/wild-type) from another (the variant/mutant/non-reference). There were perhaps 2 proposals for this [more details required…].
Tables
Table: feature
A feature is a biological sequence or a section of a biological sequence, or a collection of such sections. Examples include genes, exons, transcripts, regulatory regions, polypeptides, protein domains, chromosome sequences, sequence variations, cross-genome match regions such as hits and HSPs and so on; see the Sequence Ontology for more.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_id | serial | PRIMARY KEY | |
| dbxref_id | integer | An optional primary public stable identifier for this feature. Secondary identifiers and external dbxrefs go in the table feature_dbxref. |
|
| organism_id | integer | UNIQUE#1 NOT NULL The organism to which this feature belongs. This column is mandatory. |
|
| name | character varying(255) | The optional human-readable common name for a feature, for display purposes. |
|
| uniquename | text | UNIQUE#1 NOT NULL The unique name for a feature; may not be necessarily be particularly human-readable, although this is preferred. This name must be unique for this type of feature within this organism. |
|
| residues | text | A sequence of alphabetic characters representing biological residues (nucleic acids, amino acids). This column does not need to be manifested for all features; it is optional for features such as exons where the residues can be derived from the featureloc. It is recommended that the value for this column be manifested for features which may may non-contiguous sublocations (e.g. transcripts), since derivation at query time is non-trivial. For expressed sequence, the DNA sequence should be used rather than the RNA sequence. |
|
| seqlen | integer | The length of the residue feature. See column:residues. This column is partially redundant with the residues column, and also with featureloc. This column is required because the location may be unknown and the residue sequence may not be manifested, yet it may be desirable to store and query the length of the feature. The seqlen should always be manifested where the length of the sequence is known. |
|
| md5checksum | character(32) | The 32-character checksum of the sequence, calculated using the MD5 algorithm. This is practically guaranteed to be unique for any feature. This column thus acts as a unique identifier on the mathematical sequence. |
|
| type_id | integer | UNIQUE#1 NOT NULL A required reference to a table:cvterm giving the feature type. This will typically be a Sequence Ontology identifier. This column is thus used to subclass the feature table. |
|
| is_analysis | boolean | NOT NULL DEFAULT false Boolean indicating whether this feature is annotated or the result of an automated analysis. Analysis results also use the companalysis module. Note that the dividing line between analysis and annotation may be fuzzy, this should be determined on a per-project basis in a consistent manner. One requirement is that there should only be one non-analysis version of each wild-type gene feature in a genome, whereas the same gene feature can be predicted multiple times in different analyses. |
|
| is_obsolete | boolean | NOT NULL DEFAULT false Boolean indicating whether this feature has been obsoleted. Some chado instances may choose to simply remove the feature altogether, others may choose to keep an obsolete row in the table. |
|
| timeaccessioned | timestamp without time zone | NOT NULL DEFAULT ('now'::text)::timestamp(6) with time
zone For handling object accession or modification timestamps (as opposed to database auditing data, handled elsewhere). The expectation is that these fields would be available to software interacting with chado. |
|
| timelastmodified | timestamp without time zone | NOT NULL DEFAULT ('now'::text)::timestamp(6) with time
zone For handling object accession or modification timestamps (as opposed to database auditing data, handled elsewhere). The expectation is that these fields would be available to software interacting with chado. |
feature Structure
Tables referencing this one via Foreign Key Constraints:
Table: feature_cvterm
Associate a term from a cv with a feature, for example, GO annotation.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_cvterm_id | serial | PRIMARY KEY | |
| feature_id | integer | UNIQUE#1 NOT NULL | |
| cvterm_id | integer | UNIQUE#1 NOT NULL | |
| pub_id | integer | UNIQUE#1 NOT NULL Provenance for the annotation. Each annotation should have a single primary publication (which may be of the appropriate type for computational analyses) where more details can be found. Additional provenance dbxrefs can be attached using feature_cvterm_dbxref. |
|
| is_not | boolean | NOT NULL DEFAULT false If this is set to true, then this annotation is interpreted as a NEGATIVE annotation - i.e. the feature does NOT have the specified function, process, component, part, etc. See GO docs for more details. |
feature_cvterm Structure
Tables referencing this one via Foreign Key Constraints:
Table: feature_cvterm_dbxref
Additional dbxrefs for an association. Rows in the feature_cvterm table may be backed up by dbxrefs. For example, a feature_cvterm association that was inferred via a protein-protein interaction may be backed by by refering to the dbxref for the alternate protein. Corresponds to the WITH column in a GO gene association file (but can also be used for other analagous associations). See http://www.geneontology.org/doc/GO.annotation.shtml#file for more details.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_cvterm_dbxref_id | serial | PRIMARY KEY | |
| feature_cvterm | feature_cvterm_id | integer | UNIQUE#1 NOT NULL |
| dbxref | dbxref_id | integer | UNIQUE#1 NOT NULL |
feature_cvterm_dbxref Structure
Table: feature_cvterm_pub
Secondary pubs for an association. Each feature_cvterm association is supported by a single primary publication. Additional secondary pubs can be added using this linking table (in a GO gene association file, these corresponding to any IDs after the pipe symbol in the publications column.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_cvterm_pub_id | serial | PRIMARY KEY | |
| feature_cvterm | feature_cvterm_id | integer | UNIQUE#1 NOT NULL |
| pub | pub_id | integer | UNIQUE#1 NOT NULL |
feature_cvterm_pub Structure
Table: feature_cvtermprop
Extensible properties for feature to cvterm associations. Examples: GO evidence codes; qualifiers; metadata such as the date on which the entry was curated and the source of the association. See the featureprop table for meanings of type_id, value and rank.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_cvtermprop_id | serial | PRIMARY KEY | |
| feature_cvterm_id | integer | UNIQUE#1 NOT NULL | |
| type_id | integer | UNIQUE#1 NOT NULL The name of the property/slot is a cvterm. The meaning of the property is defined in that cvterm. cvterms may come from the OBO evidence code cv. |
|
| value | text | The value of the property, represented as text. Numeric values are converted to their text representation. This is less efficient than using native database types, but is easier to query. |
|
| rank | integer | UNIQUE#1 NOT NULL Property-Value ordering. Any feature_cvterm can have multiple values for any particular property type - these are ordered in a list using rank, counting from zero. For properties that are single-valued rather than multi-valued, the default 0 value should be used. |
feature_cvtermprop Structure
Table: feature_dbxref
Links a feature to dbxrefs. This is for secondary identifiers; primary identifiers should use feature.dbxref_id.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_dbxref_id | serial | PRIMARY KEY | |
| feature_id | integer | UNIQUE#1 NOT NULL | |
| dbxref_id | integer | UNIQUE#1 NOT NULL | |
| is_current | boolean | NOT NULL DEFAULT true True if this secondary dbxref is the most up to date accession in the corresponding db. Retired accessions should set this field to false. |
feature_dbxref Structure
Table: feature_pub
Provenance. Linking table between features and publications that mention them.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_pub_id | serial | PRIMARY KEY | |
| feature | feature_id | integer | UNIQUE#1 NOT NULL |
| pub | pub_id | integer | UNIQUE#1 NOT NULL |
feature_pub Structure
Tables referencing this one via Foreign Key Constraints:
Table: feature_pubprop
Property or attribute of a feature_pub link.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_pubprop_id | serial | PRIMARY KEY | |
| feature_pub | feature_pub_id | integer | UNIQUE#1 NOT NULL |
| cvterm | type_id | integer | UNIQUE#1 NOT NULL |
| value | text | ||
| rank | integer | UNIQUE#1 NOT NULL |
feature_pubprop Structure
Table: feature_relationship
Features can be arranged in graphs, e.g. “exon part_of transcript part_of gene”; If type is thought of as a verb, the each arc or edge makes a statement [Subject Verb Object]. The object can also be thought of as parent (containing feature), and subject as child (contained feature or subfeature). We include the relationship rank/order, because even though most of the time we can order things implicitly by sequence coordinates, we can not always do this - e.g. transpliced genes. It is also useful for quickly getting implicit introns.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_relationship_id | serial | PRIMARY KEY | |
| subject_id | integer | UNIQUE#1 NOT NULL The subject of the subj-predicate-obj sentence. This is typically the subfeature. |
|
| object_id | integer | UNIQUE#1 NOT NULL The object of the subj-predicate-obj sentence. This is typically the container feature. |
|
| type_id | integer | UNIQUE#1 NOT NULL Relationship type between subject and object. This is a cvterm, typically from the OBO relationship ontology, although other relationship types are allowed. The most common relationship type is OBO_REL:part_of. Valid relationship types are constrained by the Sequence Ontology. |
|
| value | text | Additional notes or comments. |
|
| rank | integer | UNIQUE#1 NOT NULL The ordering of subject features with respect to the object feature may be important (for example, exon ordering on a transcript - not always derivable if you take trans spliced genes into consideration). Rank is used to order these; starts from zero. |
feature_relationship Structure
Tables referencing this one via Foreign Key Constraints:
Table: feature_relationship_pub
Provenance. Attach optional evidence to a feature_relationship in the form of a publication.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_relationship_pub_id | serial | PRIMARY KEY | |
| feature_relationship | feature_relationship_id | integer | UNIQUE#1 NOT NULL |
| pub | pub_id | integer | UNIQUE#1 NOT NULL |
feature_relationship_pub Structure
Table: feature_relationshipprop
Extensible properties for feature_relationships. Analagous structure to featureprop. This table is largely optional and not used with a high frequency. Typical scenarios may be if one wishes to attach additional data to a feature_relationship - for example to say that the feature_relationship is only true in certain contexts.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_relationshipprop_id | serial | PRIMARY KEY | |
| feature_relationship_id | integer | UNIQUE#1 NOT NULL | |
| type_id | integer | UNIQUE#1 NOT NULL The name of the property/slot is a cvterm. The meaning of the property is defined in that cvterm. Currently there is no standard ontology for feature_relationship property types. |
|
| value | text | The value of the property, represented as text. Numeric values are converted to their text representation. This is less efficient than using native database types, but is easier to query. |
|
| rank | integer | UNIQUE#1 NOT NULL Property-Value ordering. Any feature_relationship can have multiple values for any particular property type - these are ordered in a list using rank, counting from zero. For properties that are single-valued rather than multi-valued, the default 0 value should be used. |
feature_relationshipprop Structure
Tables referencing this one via Foreign Key Constraints:
Table: feature_relationshipprop_pub
Provenance for feature_relationshipprop.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_relationshipprop_pub_id | serial | PRIMARY KEY | |
| feature_relationshipprop | feature_relationshipprop_id | integer | UNIQUE#1 NOT NULL |
| pub | pub_id | integer | UNIQUE#1 NOT NULL |
feature_relationshipprop_pub Structure
Table: feature_synonym
Linking table between feature and synonym.
| F-Key | Name | Type | Description |
|---|---|---|---|
| feature_synonym_id | serial | PRIMARY KEY | |
| synonym_id | integer | UNIQUE#1 NOT NULL | |
| feature_id | integer | UNIQUE#1 NOT NULL | |
| pub_id | integer | UNIQUE#1 NOT NULL The pub_id link is for relating the usage of a given synonym to the publication in which it was used. |
|
| is_current | boolean | NOT NULL DEFAULT true The is_current boolean indicates whether the linked synonym is the current -official- symbol for the linked feature. |
|
| is_internal | boolean | NOT NULL DEFAULT false Typically a synonym exists so that somebody querying the db with an obsolete name can find the object theyre looking for (under its current name. If the synonym has been used publicly and deliberately (e.g. in a paper), it may also be listed in reports as a synonym. If the synonym was not used deliberately (e.g. there was a typo which went public), then the is_internal boolean may be set to -true- so that it is known that the synonym is -internal- and should be queryable but should not be listed in reports as a valid synonym. |
feature_synonym Structure
Table: featureloc
The location of a feature relative to another feature. Important: interbase coordinates are used. This is vital as it allows us to represent zero-length features e.g. splice sites, insertion points without an awkward fuzzy system. Features typically have exactly ONE location, but this need not be the case. Some features may not be localized (e.g. a gene that has been characterized genetically but no sequence or molecular information is available). Note on multiple locations: Each feature can have 0 or more locations. Multiple locations do NOT indicate non-contiguous locations (if a feature such as a transcript has a non-contiguous location, then the subfeatures such as exons should always be manifested). Instead, multiple featurelocs for a feature designate alternate locations or grouped locations; for instance, a feature designating a blast hit or hsp will have two locations, one on the query feature, one on the subject feature. Features representing sequence variation could have alternate locations instantiated on a feature on the mutant strain. The column:rank is used to differentiate these different locations. Reflexive locations should never be stored - this is for -proper- (i.e. non-self) locations only; nothing should be located relative to itself.
| F-Key | Name | Type | Description |
|---|---|---|---|
| featureloc_id | serial | PRIMARY KEY | |
| feature_id | integer | UNIQUE#1 NOT NULL The feature that is being located. Any feature can have zero or more featurelocs. |
|
| srcfeature_id | integer | The source feature which this location is relative to. Every location is relative to another feature (however, this column is nullable, because the srcfeature may not be known). All locations are -proper- that is, nothing should be located relative to itself. No cycles are allowed in the featureloc graph. |
|
| fmin | integer | The leftmost/minimal boundary in the linear range represented by the featureloc. Sometimes (e.g. in Bioperl) this is called -start- although this is confusing because it does not necessarily represent the 5-prime coordinate. Important: This is space-based (interbase) coordinates, counting from zero. To convert this to the leftmost position in a base-oriented system (eg GFF, Bioperl), add 1 to fmin. |
|
| is_fmin_partial | boolean | NOT NULL DEFAULT false This is typically false, but may be true if the value for column:fmin is inaccurate or the leftmost part of the range is unknown/unbounded. |
|
| fmax | integer | The rightmost/maximal boundary in the linear range represented by the featureloc. Sometimes (e.g. in bioperl) this is called -end- although this is confusing because it does not necessarily represent the 3-prime coordinate. Important: This is space-based (interbase) coordinates, counting from zero. No conversion is required to go from fmax to the rightmost coordinate in a base-oriented system that counts from 1 (e.g. GFF, Bioperl). |
|
| is_fmax_partial | boolean | NOT NULL DEFAULT false This is typically false, but may be true if the value for column:fmax is inaccurate or the rightmost part of the range is unknown/unbounded. |
|
| strand | smallint | The orientation/directionality of the location. Should be 0, -1 or +1. |
|
| phase | integer | Phase of translation with respect to srcfeature_id. Values are 0, 1, 2. It may not be possible to manifest this column for some features such as exons, because the phase is dependant on the spliceform (the same exon can appear in multiple spliceforms). This column is mostly useful for predicted exons and CDSs. |
|
| residue_info | text | Alternative residues, when these differ from feature.residues. For instance, a SNP feature located on a wild and mutant protein would have different alternative residues. for alignment/similarity features, the alternative residues is used to represent the alignment string (CIGAR format). Note on variation features; even if we do not want to instantiate a mutant chromosome/contig feature, we can still represent a SNP etc with 2 locations, one (rank 0) on the genome, the other (rank 1) would have most fields null, except for alternative residues. |
|
| locgroup | integer | UNIQUE#1 NOT NULL This is used to manifest redundant, derivable extra locations for a feature. The default locgroup=0 is used for the DIRECT location of a feature. Important: most Chado users may never use featurelocs WITH logroup > 0. Transitively derived locations are indicated with locgroup > 0. For example, the position of an exon on a BAC and in global chromosome coordinates. This column is used to differentiate these groupings of locations. The default locgroup 0 is used for the main or primary location, from which the others can be derived via coordinate transformations. Another example of redundant locations is storing ORF coordinates relative to both transcript and genome. Redundant locations open the possibility of the database getting into inconsistent states; this schema gives us the flexibility of both warehouse instantiations with redundant locations (easier for querying) and management instantiations with no redundant locations. An example of using both locgroup and rank: imagine a feature indicating a conserved region between the chromosomes of two different species. We may want to keep redundant locations on both contigs and chromosomes. We would thus have 4 locations for the single conserved region feature - two distinct locgroups (contig level and chromosome level) and two distinct ranks (for the two species). |
|
| rank | integer | UNIQUE#1 NOT NULL Used when a feature has >1 location, otherwise the default rank 0 is used. Some features (e.g. blast hits and HSPs) have two locations - one on the query and one on the subject. Rank is used to differentiate these. Rank=0 is always used for the query, Rank=1 for the subject. For multiple alignments, assignment of rank is arbitrary. Rank is also used for sequence_variant features, such as SNPs. Rank=0 indicates the wildtype (or baseline) feature, Rank=1 indicates the mutant (or compared) feature. |
featureloc Structure
| Name | Constraint |
|---|---|
| featureloc_c2 | CHECK ((fmin <= fmax)) |
featureloc Constraints
Tables referencing this one via Foreign Key Constraints:
Table: featureloc_pub
Provenance of featureloc. Linking table between featurelocs and publications that mention them.
| F-Key | Name | Type | Description |
|---|---|---|---|
| featureloc_pub_id | serial | PRIMARY KEY | |
| featureloc | featureloc_id | integer | UNIQUE#1 NOT NULL |
| pub | pub_id | integer | UNIQUE#1 NOT NULL |
featureloc_pub Structure
Table: featureprop
A feature can have any number of slot-value property tags attached to it. This is an alternative to hardcoding a list of columns in the relational schema, and is completely extensible.
| F-Key | Name | Type | Description |
|---|---|---|---|
| featureprop_id | serial | PRIMARY KEY | |
| feature_id | integer | UNIQUE#1 NOT NULL | |
| type_id | integer | UNIQUE#1 NOT NULL The name of the property/slot is a cvterm. The meaning of the property is defined in that cvterm. Certain property types will only apply to certain feature types (e.g. the anticodon property will only apply to tRNA features) ; the types here come from the sequence feature property ontology. |
|
| value | text | The value of the property, represented as text. Numeric values are converted to their text representation. This is less efficient than using native database types, but is easier to query. |
|
| rank | integer | UNIQUE#1 NOT NULL Property-Value ordering. Any feature can have multiple values for any particular property type - these are ordered in a list using rank, counting from zero. For properties that are single-valued rather than multi-valued, the default 0 value should be used |
featureprop Structure
Tables referencing this one via Foreign Key Constraints:
Table: featureprop_pub
Provenance. Any featureprop assignment can optionally be supported by a publication.
| F-Key | Name | Type | Description |
|---|---|---|---|
| featureprop_pub_id | serial | PRIMARY KEY | |
| featureprop | featureprop_id | integer | UNIQUE#1 NOT NULL |
| pub | pub_id | integer | UNIQUE#1 NOT NULL |
featureprop_pub Structure
Table: synonym
A synonym for a feature. One feature can have multiple synonyms, and the same synonym can apply to multiple features.
| F-Key | Name | Type | Description |
|---|---|---|---|
| synonym_id | serial | PRIMARY KEY | |
| name | character varying(255) | UNIQUE#1 NOT NULL The synonym itself. Should be human-readable machine-searchable ascii text. |
|
| type_id | integer | UNIQUE#1 NOT NULL Types would be symbol and fullname for now. |
|
| synonym_sgml | character varying(255) | NOT NULL The fully specified synonym, with any non-ascii characters encoded in SGML. |
synonym Structure
Tables referencing this one via Foreign Key Constraints: